

Three-component cross-coupling reactions involving alkenes enabled by visible-light and earth-abundant-metal-catalysis
 0
0 
Abstract
Catalytic difunctionalizations of alkenes represent one of the most straightforward and efficient ways to build molecular complexity due to the simultaneous installation of two vicinal chemical bonds onto alkenes, providing profound chemical space for selective transformations in organic synthesis. Over the past decade, the merge of visible-light catalysis and earth-abundant-metal catalysis has taken advantage of green catalysis, thus evolving into an enabling platform for difunctionalization of alkenes. This dual catalytic mode facilitates the construction of multiple chemical bonds over the π-bond of alkenes in one step, providing a mild and straightforward method for the rapid construction of sp3-rich structures in a selective manner. In this review, we systematically summarized the progress of three-component cross-coupling reaction of olefins catalyzed by visible-light and first-row transition metal over the past decade. The combination of visible-light with different first-row transition metals, such as copper, nickel, chromium, titanium, manganese and iron, is discussed, along with the detailed reaction mechanisms. The scope of alkenes in this review includes alkenes, 1,3-dienes and 1,3-enynes. Moreover, the future directions and efforts in visible-light and earth-abundant-metal-catalyzed three-component reactions of alkenes are also discussed.
Keywords
INTRODUCTION
Transition-metal-catalyzed cross-coupling reactions have fundamentally changed the strategy in organic synthesis and are among the most efficient and straightforward ways to construct diverse chemical bonds, which have significantly enhanced the ability to access complex organic molecules and found broad applications in the synthesis of natural products and functional materials[1-5]. Over the past few decades, significant advancements have been made in noble-transition-metal-catalyzed cross-coupling reactions to form C−C and C−heteroatom bonds[6-10]. However, the use of precious metals such as platinum, palladium, and rhodium hampered their further applications due to their high cost, toxicity, and rarity[11]. In this regard, earth-abundant transition metals, such as iron, cobalt, nickel, and copper, are becoming increasingly important for the sustainable future of organic synthesis and manufacturing due to their low cost, low toxicity, and abundance[12-17]. The development of efficient methodology based on these earth-abundant transition metals offers potential synthetic methods with environmental and economic benefits and provides opportunities to discover new chemical space in bond-forming processes and synthesis.
Given the abundant and renewable nature of visible light, photochemistry and photocatalysis [via photoinduced electron transfer (PET)] have been proven to be a powerful tool for organic synthesis[18-25]. To this end, the combination of photoredox and earth-abundant transition metals offers a new and enabling platform for new methodology development, which provides great potential for the development of more sustainable and cost-effective synthetic methodologies in organic synthesis and opens up new avenues for exploring catalysis in diverse chemical spaces[26-34].
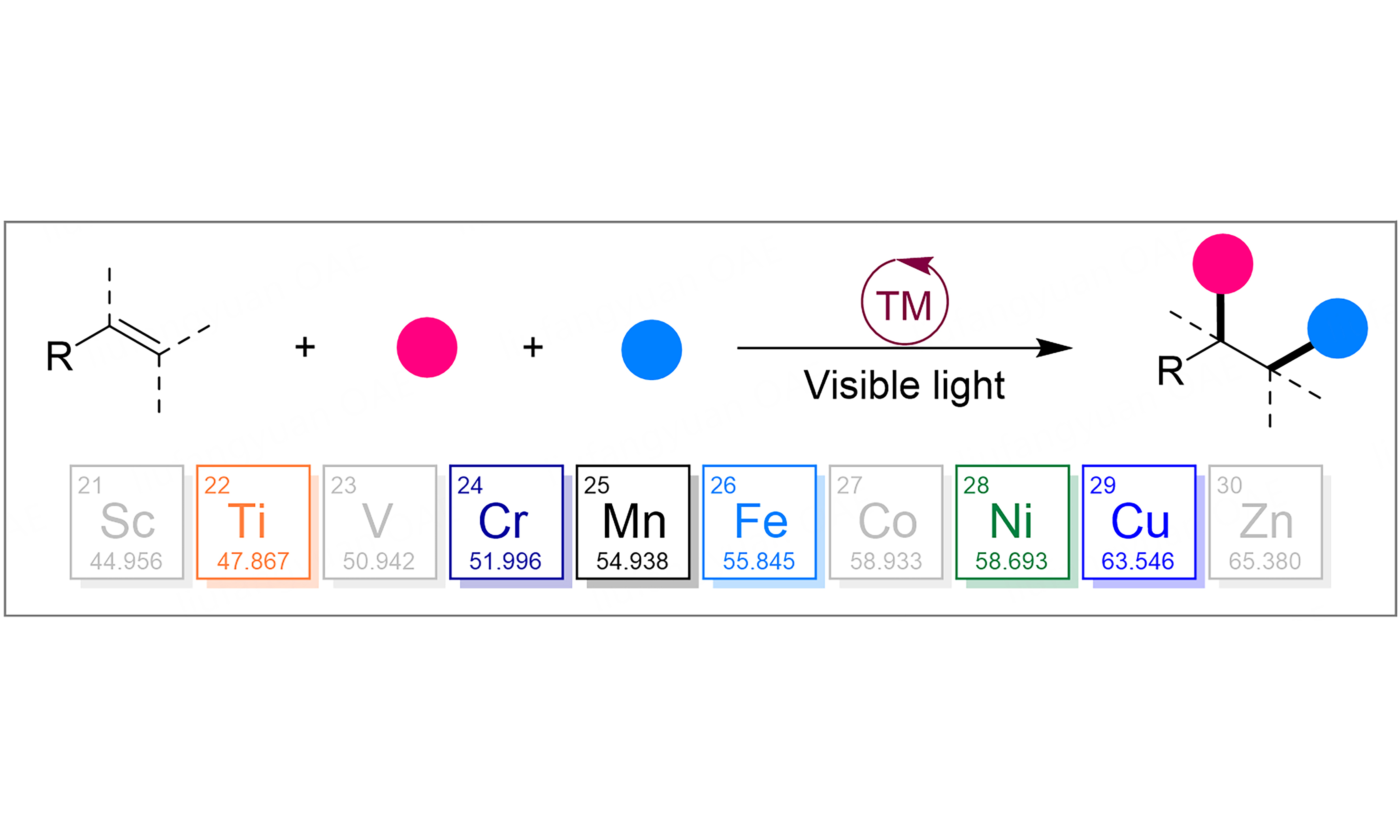
On the other hand, as abundant and readily available feedstocks, alkenes have been recently utilized as prominent platforms for the diverse transformation and precursors for other functional groups with the formation of C−Y (Y = C, N, O, X, S, etc.) bonds[35-37]. Among these, catalytic three-component difunctionalization of alkenes represents a powerful and efficient technology for the rapid construction of molecular complexity in a single operation due to the simultaneous formation of two vicinal chemical bonds[38-41]. Over the past decade, three-component difunctionalizations of alkenes enabled by the merger of photoredox and earth-abundant transition-metal catalysis have gained great attention from the community and achieved significant progress.
In this review, the progress in merging visible-light and earth-abundant-transition-metals (Cu, Ni, Cr, Ti, Mn and Fe) catalyzed three-component difunctionalizations of alkenes over recent years has been summarized. The reactions and the mechanistic aspects are discussed to provide insight into the reaction development. Moreover, further efforts and directions in this area are also discussed. In addition, the review is divided into different categories based on the types of earth-abundant transition metals.
1. Photoredox and copper-catalyzed three-component difunctionalizations of alkenes:
1.1 Three-component cross-coupling involving monoalkenes.
1.2 Three-component cross-coupling involving 1,3-diene/enyne.
2. Photoredox and nickel-catalyzed three-component difunctionalizations of alkenes.
3. Photoredox and chromium-catalyzed three-component difunctionalizations of alkenes.
4. Photoredox and titanium-catalyzed three-component difunctionalizations of alkenes.
5. Photoredox and manganese-catalyzed three-component difunctionalizations of alkenes.
6. Photoredox and iron-catalyzed three-component difunctionalizations of alkenes.
PHOTOREDOX AND COPPER-CATALYZED THREE-COMPONENT DIFUNCTIONALIZATIONS OF ALKENES
Copper is emerging as a viable transition metal for catalytic cross-coupling reactions to forge carbon-carbon and carbon-heteroatom bonds. Moreover, excited copper complexes have strong reducing ability, sufficient lifetime and high luminescence intensity, which make them ideal candidates as photosensitizers[42,43]. Accordingly, different reaction mechanisms and roles for copper could be possible in copper-catalyzed reactions under visible-light irradiation, which significantly broadens the chemical space for synthesis. In 2022, Engl et al. summarized the concept, application and opportunities of atom transfer radical addition (ATRA) of copper-photocatalytic alkenes[44]. In terms of photoredox and copper-catalyzed three-component difunctionalizations of alkenes, typical general mechanisms are proposed and depicted in Scheme 1.
The first general pathway for copper-catalyzed alkene-involved three-component cross-coupling reactions under visible-light irradiation proceeds in the absence of external photocatalysts (PCs) [Scheme 1(a)]. First, cupric salt CuX and ligand L generate intermediate 1in situ, which reacts with nucleophile 2 by ligand substitution to afford CuI-Nu adduct 3. Upon irradiation, complex 3 absorbs light energy to become the excited-state complex 4, which undergoes single electron transfer (SET) with electrophile 5 (such as alkyl halide) to deliver a carbon-centered radical 6 along with CuII-Nu species 7. Next, the radical 6 is subsequently captured by alkenes 8 to generate a new radical intermediate 9, which recombines with CuII-Nu species 7 to form CuIII species 10. Lastly, the trivalent copper species 10 undergoes reductive elimination to provide the final coupling product 11 and regenerate cupric salt CuX. In this reaction pathway, copper acts as both PC and transition-metal catalyst.

Scheme 1. Typical pathways for photoinduced copper-catalyzed three-component cross-coupling of alkenes.
The second general pathway for copper-catalyzed alkene-involved three-component cross-coupling reactions under visible-light irradiation proceeds in the presence of an external PC [Scheme 1(b)]. First, the excited-state PC* oxidizes the electrophilic reagents 5 to generate a carbon-centered radical 6 through a single-electron oxidation process, which then adds to the alkenes 8 to give new radicals 9. Then, the CuII-Nu complex 7 is intercepted by radical 9 to furnish CuIII species 10, which undergoes rapid reductive elimination to deliver final product 11 and CuI complex 1. Finally, 1 binds to the nucleophile Nu-, which is then oxidized by PC+• to regenerate CuII species 7 and ground-state PC, thereby finishing two catalytic cycles.
The above-mentioned pathways are initiated by radical generation along with the formation of CuII from CuI. The third pathway is generation of radicals by ligand-to-metal charge-transfer (LMCT) process to generate CuI from CuII [Scheme 1(c)][45-49]. Upon irradiation, LCuIINuX 7 is excited to undergo LMCT to generate radical Nu• and LCuI. Next, Nu• is intercepted by alkenes to generate a new carbon-centered radical 9’, which combines with electrophiles to afford the desired product 11 in the presence of LCuI.
Three-component cross-coupling involving monoalkenes
In 2015, Fumagalli et al. developed a photoredox copper-catalyzed three-component reaction of styrenes 12 with Zhdankin reagent 13 and nucleophile 14 (ROH or NaBr) to afford nitrogen-containing small molecule building blocks 15 in 34%-96% yields [Scheme 2][50]. This transformation generates azide radicals from 13 under photocatalytic conditions, followed by the addition of alkenes 12 to obtain carbon-centered radicals, which are ultimately captured by polar molecules to deliver the final product 15. Notably, this catalytic system is applicable only to aromatic alkenes as radical acceptors and methanol, trifluoroethanol, and sodium bromide as nucleophiles.

Scheme 2. Three-component azidation of styrenes with Zhdankin reagent and nucleophiles.
Moreover, [Cu(dap)2Cl] can mediate the LMCT pathway. In 2018, Hossain et al. developed a photoinduced Cu-catalyzed oxo-azidation of arylalkenes with TMSN3 and O2 to give α-N3-substituted aromatic ketones[51]. The reaction undergoes a LMCT process to generate radicals [Scheme 1(c)], where N3• is captured by styrenes to generate benzyl radicals. Interception of benzylic radicals by O2 to produce the final products. In 2023, He et al. reported a visible-light-induced copper-catalyzed three-component reaction of arylalkenes with MgCl2 and O2 to deliver α-chloroketones[52].
In 2017, Guo et al. reported the first example of photoinduced, copper-catalyzed three-component cyanofluoroalkylation of alkenes using fluoroalkyl iodides 16 as fluoroalkylation reagents [Scheme 3][53]. With a compact fluorescent light bulb irradiation, alkenes 8 react with fluoroalkyl iodides 16 and trimethylsilyl cyanide (TMSCN) 17 to give difunctionalization product 18 in 48%-91% yields. The strategy uses N,N-diisopropylethylamine (DIPEA) as a sacrificial reducing agent, and is applicable to both alkyl and aryl alkenes. Moreover, using inexpensive and readily available fluorinated alkyl iodides instead of Togni’s or Umemoto’s reagents as fluoroalkyl precursors renders the reaction suitable for large-scale synthesis.

Scheme 3. Photoinduced and copper-catalyzed cyanofluoroalkylation of alkenes.
Copper acetate [Cu(OAc)2] and extraneous cyanide TMSCN 17 undergo rapid ligand exchange and are followed by SET with electron-rich DIPEA to form CuICN 19, which is converted into its triplet state
In 2018, He et al. described a visible-light-promoted CuI/BINAP catalyzed three-component cross-coupling of alkenes (including styrenes and electron-poor alkenes) 8 with alkyl halides 25 and nucleophiles 26a (trifluoromethylthiolate, bromide, cyanide and azide) to afford 27 in 54%-98% yields [Scheme 4(a)][54]. In this process, a photoexcited LCuINu complex served as a reductant to generate an alkyl radical from alkyl halides. Moreover, this catalytic system is suitable for styrenes or electron-poor alkenes, but the alkyl halides require strong electron-withdrawing groups at β-position, such as carbonyl, fluorine or sulfone groups.

Scheme 4. Visible-light-induced, copper-catalyzed three-component cross-coupling of alkenes with alkyl halides and nucleophiles.
In 2019, Xiong et al. reported a visible-light-mediated copper-catalyzed carboamination of alkenes with alkyl halides and primary/secondary amines to give alkylamination with 30%-86% yields [Scheme 4(b)][55]. In this reaction, CuCl serves as both PC and cross-coupling catalyst. This method is suitable for aryl alkenes and 1,3-dienes and tolerates both primary and secondary amines under mild conditions. Notably, a catalytic amount of ligand (rac-BINOL) is needed for this transformation when non-carbazole secondary amines are used as coupling partners.
In 2023, Hu et al. reported a photoinduced and Cu-catalyzed three-component cross-coupling reaction of styrenes with KSCN and fluorinated alkyl halide to give difluorothiocyanate compounds in 41%-82% yields [Scheme 4(c)][56]. CuI/rac-BINAP plays a dual role as photoredox catalyst for electron transfer and cross-coupling catalyst for C−SCN bond formation. Notably, this catalytic system is compatible with various 1-, 1,2-, 1,1-, and 1,1,2-substituted styrenes and directed aliphatic alkenes. Moreover, activated alkyl halides are suitable alkyl radical precursors for this reaction.
Similarly, Zhang et al. reported a photoinduced, Cu-catalyzed three-component cross-coupling of aryl alkenes with terminal alkynes and aryl/alkyl iodides to form propargylic skeleton in 40%-93% yields[57]. This method uses 2,2’:6’,2”-terpyridine as a ligand and is applicable to hetero/aryl alkenes, terminal aryl/alkylalkynes, and aryl/alkyl iodides. Later, Zhang et al. developed visible-light-mediated Cu-catalyzed enantioselective alkylalkynylation and arylalkynylation of styrenes with terminal alkynes and alkyl/aryl iodides to afford chiral propargylic building blocks in 13%-87% yields with up to 98% ee [Scheme 4(d)][58]. Chiral CuI/tBu-BOPA complexes act as both PC and transition-metal catalysts to induce the enantioselectivity of the products. This method is compatible with styrenes with diverse substitution patterns, terminal alkynes, and aryl and alkyl iodides. Unfortunately, aliphatic alkenes fail to undergo this enantioselective process.
The general mechanism of the reaction between alkenes, alkyl halides, and nucleophiles is described in Scheme 4. Firstly, the CuI complex 28 underwent ligand exchange with nucleophiles 26 to form intermediate CuI-nucleophile complex 29, which was converted to excited-state CuI species 30 under light radiation. Then, alkyl halides 25 interacted with 30 by SET to generate CuII-nucleophile complex 31 and alkyl radicals 32. Alkyl radicals 32 were captured by alkenes 8 to form a new radical species 33. Two possible pathways may lead to the formation of the target products 27 from 33. Radicals 33 rebounded with CuII species 31 to generate CuIII complex 34, followed by reductive elimination to provide the products 27 and regenerate CuI species 28 (path a). Alternatively, radicals 33 could directly coupling with 31 through a SET process to deliver the target products 27 and CuI species 28 (path b).
In 2018, Yu et al. developed a visible-light-promoted Cu/dtbbpy catalyzed three-component cross-coupling of styrenes 12 with oxime esters 35 and nucleophiles aryl/vinyl boronic acids 26e, providing 1,1-diarylmethane containing alkylnitriles 36 in 20%-90% yields [Scheme 5(a)][59]. This protocol is applicable to various aromatic alkenes, aryl and vinyl boronic acids. However, only oxime esters derived from cyclobutanones are suitable for this reaction. Very recently, a general visible-light-promoted copper-catalyzed asymmetric three-component cross-coupling of alkenes with oxime carbonates and aryl boronic acids has been developed, delivering chiral benzylic alkynes and allenes in 19%-87% yields with 30%-96% ee[60]. Notably, copper/Box complexes play a dual role in both alkyl radical generation and subsequent asymmetric cross-coupling with aryl boronic acids.

Scheme 5. Photoinduced copper-catalyzed cross-coupling of styrenes with oxime esters and nucleophiles.
Moreover, using terminal alkynes 26d instead of arylboronic acids, reaction can be conducted under similar conditions to afford alkylalkynylation of styrenes in 14%-76% yields [Scheme 5(b)][61]. This strategy combines photoinduced iminyl radical mediated C−C bond cleavage with copper-catalyzed C(sp3)−C(sp) cross-coupling.
These reactions utilized oxime esters as cyanoalkyl radical precursors, which underwent SET with ligated CuI species to give CuII species and iminyl radicals 37, followed by ring-opening of four-membered ring to give cyanoalkyl radicals 38. Radical addition of 38 onto styrenes 12 generated benzylic radicals 39. Recombination of 39 with CuII-Nu species 40, generated from transmetalation of ligated CuII species with nucleophiles 26 (aryl boronic acids or terminal alkynes), gave CuIII species 41. Reductive elimination of 41 delivered the final products and the regeneration of ligated CuI species.
In 2019, Lv et al. developed a visible-light-promoted copper-catalyzed three-component coupling of alkenes 8 with aryl boronic acids 26e and α-bromocarbonyl precursors 42 to synthesize γ arylated carbonyls 43 in 45%-88% yields [Scheme 6][62]. In the absence of a directing group, the protocol allows for the sequential formation of C(sp3)−C(sp3) and C(sp3)−C(sp2) bonds under ambient conditions. Additionally, this catalytic method is compatible with a wide range of aryl and alkyl alkenes, aryl boronic acids, and α-bromo-carbonyls.

Scheme 6. Photoinduced and copper-catalyzed radical relay alkylarylation of alkenes.
Under visible-light irradiation, the ground-state photocatalyst IrIII was converted into its excited-state IrIII*, which interacted with α-bromocarbonyl precursor 42 by SET process to afford an alkyl radical 44 and IrIV. Then, intermediate 44 was trapped by alkene 8 to give a new alkyl radical 45. Meanwhile, CuI transmetalated with aryl boronic acid 26e in the presence of a base to provide Ar-CuI intermediate 46. Rebound of 46 with alkyl radical 45 formed CuII intermediate 47, which further underwent SET to reduce IrIV to ground-state IrIII, followed by reductive elimination to deliver the final products 43 and CuI species to close the catalytic cycles.
Then, Lv et al. further described a visible-light and copper dual-catalyzed three-component cross-coupling of alkenes 8 with α-bromocarbonyl precursors 42 and terminal alkyne silicates 48 to furnish γ-alkynylated esters, acids, amides, ketones and aldehydes 49 in 33%-80% yields [Scheme 7][63]. The strategy is operationally simple and does not require a directing group, and a series of readily available starting materials can be assembled at the γ-position of the carbonyl group. CuIL complex 50 coordinates with alkynes to form alkynyl CuI species 51, which interacts with α-bromocarbonyl precursors 42 to give alkyl radicals 52 and alkynyl CuII53. Then, alkyl radicals add to the C=C double bond of alkenes 8 to provide benzyl radicals 54, which are then intercepted by 53 to afford key intermediate CuIII species 55. Finally, CuIII species 55 are excited by IrIII photocatalyst to generate [CuIII]* species 56, followed by accelerated reductive elimination to produce desired products 49.

Scheme 7. Visible-light-induced copper-catalyzed alkylalkynylation of styrenes.
In 2021, Wang et al. developed a photoredox and copper-catalyzed three-component enantioselective carbocyanation of styrenes 12 with redox-active oxime esters (57 or 35) and TMSCN 17 to provide β-cyano ketones (58) and 1,5-dinitriles (59) in 24%-86% yields with 82%-97% ee [Scheme 8][64]. The success of this process hinges on the chemoselective addition of acyl and cyanoalkyl radicals to styrenes, which are generated in situ by the C−C bond cleavage triggered by the photocatalytic nitrogen-centered iminyl radical of the redox-active oxime ester. This reaction tolerates a range of styrenes, yet fails to aliphatic alkenes.

Scheme 8. Photoinduced Cu-catalyzed asymmetric three-component carbocyanation of styrenes.
The reaction is initiated by the excitation of the PC to PC* under purple light-emitting diode (LED) irradiation. Redox-active oxime esters 57 or 35 went through a SET reduction process with PC* to afford iminyl radicals 60 or 37 along with carboxylic anions and oxidized photocatalyst PC•+. Next, iminyl radicals underwent C−C bond β-cleavage to form acyl and cyanoalkyl radicals 61 and 38, which were trapped by styrenes 12 through radical addition to deliver benzylic radicals 62. Meanwhile, initially released carboxylic anion promoted the ligand exchange between LCuI species and TMSCN 17 to furnish LCuICN complex 63, which interacted with PC•+ to generate LCuII(CN)2 complex 64 and regenerate PC in the presence of TMSCN. Next, pro-chiral benzylic radicals 62 rebounded with 64 to form chiral high-valent CuIII complex 65, followed by reductive elimination to give enantioenriched β-cyano ketones 58 or alkyldinitriles 59 and regenerate CuI species 63.
As a class of zwitterions, the transformation of sulfur ylides through ion pathways has been widely studied. However, there are relatively few reports on sulfur ylides as radical precursors. In 2022, Yan et al. developed a photoinduced and stoichiometric Cu(TFA)2·H2O mediated three-component radical addition/hydroxylation reaction of styrenes 12 with sulfur ylides 66 to deliver γ-hydroxy carbonyl compounds 67 in 44%-99% yields in the presence of water [Scheme 9][65]. α-Carbonyl carbon-centered radicals 70 were the key intermediate in the transformation, which was formed by the proton-coupled electron transfer (PCET) of sulfur ylides 66 in a mixed solvent [CF3CH2OH/dichloromethane (DCM) = 4:1] via 68 to release dimethyl sulfide 69. Addition of 70 to styrenes 12 led to the formation of benzylic radicals 71, which was oxidized to benzylic cations 72 enabled by stoichiometric Cu(TFA)2·H2O. In the presence of water, 72 was quenched to deliver the final product 67. Moreover, this protocol is suitable for styrenes, yet not for aliphatic alkenes.

Scheme 9. Visible-light and copper mediated alkylhydroxylation of styrenes.
Atom transfer radical addition (ATRA) reaction or Kharasch addition was first discovered by Kharasch et al. in the 1940s[66-68]. In 2022,

Scheme 10. Photoinduced Cu-catalyzed enantioselective alkylesterificaiton of styrenes.
The transformation proceeds under redox-neutral conditions without the use of external oxidants. First, the ligand exchange between the complex of Cu(CH3CN)4PF6/(S,S)-L and carboxylic acids 73 provided light-absorbing intermediate [L/CuI/X] (X = PF6- or ArCO2-) species 76. Upon purple light irradiation, 76 was excited to its excited-state [L/CuI/X]*77, followed by SET with oxime esters 35 to furnish iminyl radicals 37 and [LCuII/O2CAr]X 78 along with the formation of carboxylic anion (BocO-). Driven ring strain release, 37 underwent β−C−C bond cleavage to afford cyanoalkyl radicals 38, which further added to styrenes 12 to form benzylic radicals 79. Subsequently, 78 recombined with benzylic radicals 79 to provide CuIII species 80 via a radical rebound process. Finally, intermediate 80 went through reductive elimination to forge enantioselective C−O bond to give final products 75 and regenerate 76.
To date, synthesis of highly functionalized acyclic (hetero)arylethylamines remains challenging. Cai et al. described a visible-light-induced and copper-catalyzed three-component reaction of alkenes 8 with arylthianthrenium salts 81 and sodium azide 82 to deliver functionalized acyclic (hetero)arylethylamine scaffolds 83 in 36%-73% yields [Scheme 11][70]. Importantly, rac-BINAP coordinated with Cu(CH3CN)4BF4 and NaN3in situ to form rac-BINAP-CuI-N384 as photoactive catalytic species, and its structure was confirmed by X-ray diffraction analysis. Notably, this strategy can be applied to both aryl and aliphatic alkenes.

Scheme 11. Photoinduced copper-catalyzed arylazidation of alkenes.
Upon visible-light irradiation, rac-BINAP-CuI-azide 84 was converted to its excited-state 85, followed by reaction with arylthianthrenium salts 81 by single-electron reduction to form aryl radicals 86 and CuII complex 87 or its cationic counterpart. Next, aryl radicals 86 were captured by carbon-carbon double bonds of alkenes 8 to produce alkyl radicals 88, which underwent azido transfer from 87 via an outer-sphere route to afford the products 83 and regenerate photoactive catalytic species 84.
In 2024, Zhang et al. developed a visible-light-promoted and copper-catalyzed enantioselective cyanofunctionalization of styrenes 12 with electrophiles (H+, D+, CO2) 89 and nucleophiles 90 to deliver a series of chiral cyanides 91 with 23%-97% yields and 64%-99% ee [Scheme 12][71]. This dual catalytic strategy generates alkene radical anions, achieving regio-, chemo-, and enantioselective hydrocyanation, deuterocyanation and cyanocarboxylation of styrenes. Additionally, this method is suitable for various terminal and internal styrenes under mild conditions.

Scheme 12. Photoinduced copper-catalyzed enantioselective cyanofunctionalization of styrenes.
The catalytic cycle begins with the excitation of PC to PC*, which interacted with styrenes 12 by a SET process to produce the key intermediate alkene radical anion 92 and oxidized-state PC•+. Subsequently, protonation of alkene radical anion 92 by 89 (such as H2O or HCN, which was generated in situ from TMSCN and H2O) delivered benzylic radicals 93. Meanwhile, CuTc coordinated with ligand and underwent further ligand exchange with TMSCN to form the LCuICN species 94, followed by a SET pathway with oxidized PC•+ to provide LCuII(CN)2 species 95 and PC. Finally, benzylic radical 93 rebounded with 95 to give key CuIII complex 96. Further reductive elimination of 96 furnished the final products 91 and catalytic species 94.
Three-component cross-coupling involving 1,3-diene/enyne
Compounds 1,3-dienes are widely used in the synthesis of various value-added architectures. There are two reaction modes: 1,2- and 1,4-difunctionalizations of 1,3-dienes via allyl intermediates. Recently, the use of 1,3-dienes to build molecular complexity has been studied extensively. In 2021, Wang et al. reported a visible-light-mediated and Cu-catalyzed three-component coupling reaction of 1,3-dienes 97a with oxime esters 35 and carboxylic acids 73 to furnish a series of chiral esters in 63%-89% yields with 37%-96% ee under mild conditions [Scheme 13(a)][72]. Importantly, this protocol can be applied to Kharasch-Sosnovsky reaction. Moreover, this strategy was featured with mild reaction conditions, operational simplicity, inexpensive substrates, and compatibility with aryl 1,3-dienes, aryl/alkyl carboxylic acids, and cyclobutanone-type oxime esters. Unfortunately, the catalytic system has poor regioselectivity (rr = 1:1) and enantioselectivity (39% ee) for alkyl 1,3-dienes.

Scheme 13. Photoinduced copper-catalyzed asymmetric 1,2-alkylesterification of 1,3-dienes.
Building blocks, 1,3-enynes, can quickly and efficiently construct complex and functionalized molecules. The presence of both carbon-carbon double bonds and carbon-carbon triple bonds in 1,3-enynes facilitates their use in the synthesis of 1,3-dienes, allenes and propargyl compounds. Therefore, the challenge in organic synthesis is to accurately control chemo-, regio- and enantioselectivity when 1,3-enynes are involved. In 2023, Li et al. developed a visible-light-promoted Cu-catalyzed regio- and enantioselective radical coupling of 1,3-enynes 97b with oxime esters 35 and carboxylic acids 73 to form cyanoalkylated propargylic esters with 61%-95% yields and 18%-99% ee [Scheme 13(b)][73]. This protocol is compatible with a range of aryl-substituted 1,3-enyne, aryl/alkyl carboxylic acids, and oxime esters derived from cyclobutanone. However, it should be noted that the enantioselectivity of alkyl-substituted 1,3-enyne is significantly reduced.
The radical-involved multi-component bifunctionalization of 1,3-dienes has recently emerged as a promising strategy for the rapid synthesis of valuable allyl compounds in one-pot. However, expanding the scope of accessible radicals and achieving precise enantiomeric control remain major challenges. In 2024,
The general mechanism of visible-light-induced Cu-catalyzed reactions of 1,3-diene/1,3-enynes with carboxylic acids and radical precursors is illustrated in Scheme 13. The catalytic cycle starts with ligand exchange between carboxylic acids 73 and Cu(CH3CN)4PF6/L to generate [L/CuIO2CAr] species 100, which transforms to its excited-state 101 under purple light irradiation, followed by engaging in SET pathway with oxime esters 35 or Ts-ABZ 98 to give iminyl radicals 37 or azidyl radical 102, CuII species 103 and a carboxylic anion (BocO-). Subsequently, iminyl radicals 37 undergo a ring strain-driven β–C–C bond cleavage, resulting in the formation of cyanoalkyl radicals 38, which combine with 1,3-dienes 97a to provide the relatively stable allylic radicals 104. On the other hand, azidyl radical 102 is captured by 1,3-enynes 97b to form stabilized propargyl radicals 105. Then, 104 or 105 are intercepted by CuII species 103 via a radical rebound process, resulting in the formation of π-allyl CuIII complex 106. This complex undergoes reductive elimination, leading to the generation of the final product 99 in an enantioselective manner, while simultaneously regenerating 100 and completing the catalytic cycle. Notably, π-allyl CuIII complex 106 also might exist in equilibrium with hybrid allylic CuII radical 106’.
The emergence of bifunctional reagents that serve as dual coupling partners with an activating species has become valuable synthetic tools in organic chemistry. Such reagents permit the development of various reaction modes with enhanced efficiency and structural variability, which is highly sought after for atom-economic and sustainable synthesis. Chen et al. reported a visible-light-promoted copper-catalyzed regio- and enantioselective 1,2-dicarbofunctionalization of 1,3-dienes 97a with oxime esters 35 to simultaneously construct C−C and C−O bonds to deliver allylic esters in 24%-94% yields with 17%-95% ee [Scheme 14][75]. As a bifunctional reagent, the redox oxime esters form a π-allyl copper complex with 1,3-dienes 97a through a radical polarity crossing process. Furthermore, the copper-based chiral catalyst functions as both the photoredox catalyst for radical formation and the source of asymmetric induction in C−O coupling. This strategy is applicable to aryl 1,3-dienes under mild conditions. However, the regioselectivity and enantioselectivity of alkyl-substituted 1,3-dienes are poor.

Scheme 14. Photoinduced copper-catalyzed asymmetric 1,2-alkylesterification of 1,3-dienes using oxime esters as a difunctional reagent.
The process begins with Cu(CH3CN)4PF6 coordinating with (R,R)-L to generate LCuIX 108, which is excited by purple light resulting in the formation of [LCuIX]*109. Similarly, through a SET process, redox-active oximes 35 react with 109 to yield iminyl radicals 37 and CuII-nucleophile complex 110. Notably, CuII complex 110 is derived from 109 and carboxylic anion released by oxime esters. Then, iminyl radicals 37 undergo β−C−C bond cleavage and radical addition with 1,3-dienes 97a to furnish allyl radical species 111, which are captured by intermediate 110 to generate π-allyl CuIII complex 112. Finally, the reductive elimination of π-allyl CuIII complex 112 provides allylic esters 107 and regenerates 108.
Lu et al. reported a visible-light-mediated copper-catalyzed three-component enantioselective carbocyanation of 1,3-dienes 97a with N-hydroxyphthalimide (NHPI) esters 113 and TMSCN 17, yielding chiral allyl cyanides in 33%-79% yields with 7%-97% ee [Scheme 15][76]. The conditions are applicable to aromatic substituted 1,3-dienes. Unfortunately, the yield and enantioselectivity of alkyl-substituted 1,3-diene substrates are unoptimized.

Scheme 15. Photoinduced copper-catalyzed asymmetric 1,2-carbocyanation of 1,3-dienes.
A possible reaction mechanism is proposed based on control experiments. Firstly, the PC perylene is transformed into PC* under visible-light irradiation. The redox-active NHPI esters 113 are reduced by PC* to give alkyl radicals 115 and PC•+, by releasing CO2 and NHPI-. Subsequently, the alkyl radicals 115 are trapped by 1,3-dienes 97a, forming allyl radicals 116. Additionally, NHPI- interacts with TMSCN 17 to generate cyano radical 117, which coordinates with the chiral L*CuI118 complex to form the intermediate L*CuI-CN 119. Next, PC•+ oxidizes 119 to form the L*CuII-CN species 120 and PC. Finally, the allyl radicals 116 are intercepted by 120, resulting in the formation of the pivotal chiral allyl CuIII complex 121. This complex then undergoes a rapid reductive elimination to generate chiral allyl cyanides 114 and regenerate 118.
N-Aminopyridinium salts have been demonstrated as nitrogen-centered radical precursors in cross-coupling reactions. In 2022, Wu et al. reported a photoredox and Cu(CH3CN)4PF6 catalyzed regioselective three-component reaction of 1,3-dienes 97a with N-aminopyridinium salts 122 and alcohols 123 to deliver 1,2-aminoalkoxylation products 124 in 23%-71% yields [Scheme 16][77]. In this process, N-aminopyridine salts 122 act as the precursor of nitrogen-centered radicals, and alcohols 123 act as oxygen nucleophiles. This catalytic system is compatible with a series of aryl-substituted 1,3-dienes and aliphatic alcohols.

Scheme 16. Photoinduced copper-catalyzed 1,2-aminoalkoxylation of 1,3-dienes.
Under purple lights irradiation, the ground-state 10-phenylphenothiazine (Ph-PTZ) is excited to Ph-PTZ*
The light-driven, earth-abundant-metal-catalyzed 1,2-selective three-component cross-coupling reaction of 1,3-dienes has been widely reported, whereas the 1,4-addition reaction has been less frequently reported. In 2022, Bi et al. continued to report a photoinduced copper-catalyzed three-component selective

Scheme 17. Photoinduced copper-catalyzed 1,4-difluoroalkylesterification of 1,3-dienes.
In 2023, Liu et al. reported a visible-light-promoted Cu(CH3CN)4PF6 catalyzed three-component reaction of 1,3-dienes 97a with carboxylic acids 73 and N-aminopyridinium tetrafluoroborate salts 122, which delivered a series of allyl carboxylic esters and tertiary ethers 134 with yields ranging from 36% to 94% [Scheme 18][79]. Upon light irradiation, copper catalysts exhibit a dual effect, acting as PC to generate nitrogen radicals and reacting with allyl radicals through a SET process. Notably, this strategy effectively constructs a series of tertiary carbon centers. This reaction can function with diverse substrates, including a series of aryl/alkyl carboxylic acids and alkyl substituted 1,3-dienes.

Scheme 18. Photoinduced copper-catalyzed 1,2-aminoesterification of 1,3-dienes.
The mechanism investigation shows that the catalytic cycle begins when ground-state LCuI135 is excited by visible-light to 136, which interacts with N-aminopyridinium tetrafluoroborate salts 122 via SET process to deliver nitrogen-centered radicals 137 and LCuII species 138. Then, radical 137 is trapped by isoprene 97a to furnish a stable allyl radical 139, which combines with 138 by another SET approach to form allyl carbocations 140, followed by undergoing a nucleophilic attack with the carboxylic acids 73 to obtain final products 134 and regenerate ground-state LCuI135.
PHOTOREDOX AND NICKEL-CATALYZED THREE-COMPONENT DIFUNCTIONALIZATIONS OF ALKENES
As one of the earth-abundant metals, nickel has attracted considerable attention from the chemical research community due to its favorable cost, low toxicity and distinctive catalytic activity. Nickel metal has rich valence states (Ni0/NiI/NiII/NiIII/NiIV) and high energy barriers of β-hydrogen elimination, making it a promising candidate for cross-coupling reactions[40,80]. The combination of photoredox and nickel catalysis has further accelerated the development of nickel-catalyzed cross-coupling reactions, enabling challenging transformations that are difficult to achieve in a single catalytic cycle under mild reaction conditions.The three-component cross-coupling reaction of alkenes catalyzed by dual visible-light and nickel has also been the subject of extensive investigation over the past decade.
While there may be discrepancies in the specifics, the overarching mechanism of the three-component cross-coupling reaction of alkenes synergistically catalyzed by photoredox and nickel can be summarized as illustrated in Scheme 19. In the visible-light irradiation, the ground-state PC is converted to PC*, which then reacts with coupling substrate 141 by SET process to generate radical 142 and reduced PC•-. Then, radical 142 is trapped by alkene 8 by radical addition to deliver carbon-centered radicals 143, which are intercepted by Ni0 species 144 to give NiI intermediate 145, followed by oxidative addition with another coupling substrate 146 to afford NiIII complex 147. Subsequently, the NiIII intermediate 147 undergoes reductive elimination, resulting in the formation of the final product 148 and the generation of the NiI species 149. Ultimately, the reduced PC•- reduces 149 to 144 via another SET, thereby regenerating the ground-state PC and closing two catalytic cycles.

Scheme 19. Possible pathway for photoinduced and nickel-catalyzed three-component difunctionalization of alkenes.
In 2019, García-Domínguez et al. reported a visible-light-induced and nickel-catalyzed three-component difunctionalization reaction of conjugated alkenes 150 with aryl halides 151 and nucleophiles 152 to provide desired product 153 in 15%-74% yields [Scheme 20][81]. The process does not contain stoichiometric additives, the coupling agent is easy to handle, and the operation is simple. Furthermore, conjugated alkenes encompass acrylic esters, acrylonitrile, and other related compounds. Nucleophilic reagents include alkylsilicates and sulfinates, which are responsible for the production of secondary and tertiary alkyl and sulfonyl radicals, respectively.

Scheme 20. Photoredox/nickel-catalyzed alkyl/sulfonylarylation of alkenes.
The reaction begins when the ground-state PC [Ru(bpy)3]2+ is excited to an excited-state [Ru(bpy)3]2+* by visible-light, which oxidizes the nucleophiles 152 to afford the radicals 153. These are subsequently trapped by conjugated alkenes 150, thereby providing the secondary carbon-centered radical 154. Meanwhile, NiI is reduced by [Ru(bpy)3]+ to obtain Ni0, which reacts with radical 154 to generate alkyl-NiI complex 155. This is followed by oxidative addition with the aromatic halides 151, resulting in the formation of the Ar-NiIII-alkyl species 156 (path a). Alternatively, the intermediate 156 can be formed by Ni0 oxidative addition with aromatic halides 151, followed by radical recombination with 154 (path b). Finally, Ar-NiIII-alkyl species 156 undergo reductive elimination to form the final products 153 and NiI complex. The next step of the two catalytic cycles is similar, both involving the oxidation of RuI species by NiI, which regenerates the ground-state PC [Ru(bpy)3]2+ and the Ni0 catalyst, respectively.
The reaction of organoboron compounds exhibits tunable reactivity, ease of handling, and product stability, rendering it an optimal synthon for the construction of molecular complexity. In 2019, Campbell et al. described a Ni/photoredox dual catalyzed cross-coupling of vinyl boronic esters 157 with trifluoroborates 158 and aryl bromides 159 to afford α-arylboronic acid esters 160 in 35%-98% yields [Scheme 21][82]. The rapid addition of 3° radicals to alkenes and their reluctance toward single-electron metalation to Ni complexes represent a significant advancement in the field of regioselective alkylation and arylation of alkenes. Furthermore, this catalytic system is compatible with a range of aryl bromides and secondary/tertiary radical precursors, facilitating the formation of a series of tertiary/quaternary carbon centers.

Scheme 21. Three-component dicarbofunctionalization of vinyl boronates by nickel/photoredox catalysis.
The general mechanism of the reaction process is given by the authors. Upon exposure to visible-light, PC is converted to PC*, which then reacts with trifluoroborates 158 via reductive quenching by SET process, forming radicals 161. These radicals then engage in a Giese-type addition with vinyl boronic esters 157, generating radicals 162. Subsequently, the single-electron metalation of radicals 162 to Ni0 species 163 results in the formation of an alkyl-NiI complex 164, which then undergoes oxidative addition with aryl bromides 159 to yield a NiIII complex 165. Reductive elimination produces the final products 160 and NiI species 166. Lastly, the NiI species 166 undergoes SET reduction by PC-, thereby regenerating the ground-state PC and the Ni0 species 163, thus closing the two catalytic cycles. It is noteworthy that this process depends on the selective addition of radicals 161 to alkenes 157, rather than direct cross-coupling to Ni0 species 163 through single-electron metalation.
In 2020, Mega et al. developed a photoredox nickel-catalyzed decarboxylative conjunctive cross-coupling of vinyl boronic esters 167 with alkyl carboxylic acids 73 and aryl iodides 168[83]. This process afforded alkyl boronic esters 169 in yields ranging from 11% to 87% [Scheme 22]. This protocol is applicable to a range of radical precursors and aryl iodides under mild conditions. It is important to note that this transformation process presents two significant challenges. Firstly, the rate of radical 170 addition to vinyl borates 167 must exceed its rate of addition to the NiII complex 172 to avoid the formation of the two-component coupling product. This is challenging as the rate of alkyl radical addition to vinyl borates 167 is 20 times lower than to acrylate. Secondly, the reaction rate of α-boryl radicals 171 and 172 must be faster than the SET between 171 and the PC•-. This is necessary to prevent the formation of the Giese product.

Scheme 22. Photoredox/nickel-catalyzed decarboxylative alkylarylation of vinyl boronates.
Mechanism exploration indicates that the catalytic cycle commences when PC is excited to PC* under 40 W blue LED irradiation. Concurrently, the carboxylic acids 73 are converted into their conjugated bases in the presence of Cs2CO3, which interact with PC* through a SET pathway and decarboxylate to give nucleophilic
In 2020, Sun et al. reported a visible-light-promoted and nickel dual catalyzed chemo- and site-selective cross-electrophile coupling reaction of vinyl boronates 157 with aryl bromides 159 and alkyl bromides 175, which resulted in the delivery of organoboron compounds in 50%-81% yields [Scheme 23][84]. In essence, this strategy demonstrates the viability of reductive cross-coupling involving three distinct electrophilic partners in a synergistic manner. In addition to vinyl boronic acid esters, this system is compatible with other conjugated alkenes, including acrylamide, acrylic esters, and vinyl phosphate esters. Moreover, aryl bromides also have a wide range of potential substrates, although radical precursors are limited to tertiary substituted alkyl bromides.

Scheme 23. Photoredox/nickel-catalyzed 1,2-alkylarylation of vinyl boronates.
The alkyl bromides 175 and the excited-state photocatalyst PC•+ facilitate the generation of nucleophilic alkyl radicals through the SET process. These radicals then add to the vinyl borate ester 157, providing intermediates 177. It is of particular significance to note that the stability of the intermediates 177 is contingent upon the effective delocalization of the radical through the vacant p-orbital of the boron atom. Concurrently, oxidative addition of aryl bromides 159 to LNi0 results in electrophilics NiII entity 178. The recombination of 177 with 178, which occurs in a polarity-matched manner, results in the formation of the NiIII species 179. This is followed by a reductive elimination route, ultimately leading to the generation of the targeted product 176 and the NiI intermediate. Lastly, the SET pathway between NiI species and PC•- results in the formation of the ground-state PC and LNi0 species.
In 2019, Guo et al. described a photoredox/nickel dual-catalyzed three-component regioselective

Scheme 24. Photoredox/nickel-catalyzed decarboxylative alkylarylation of alkenes.
A plausible catalytic cycle of the three-component reaction is illustrated in Scheme 24. Initially, photoexcited catalyst *IrIII oxidizes oxalate 180, resulting in the generation of tertiary alkyl radicals 182 and the release of two CO2 molecules. The former is subsequently added to alkenes 8, forming nucleophilic secondary alkyl radicals 183. These radicals are then captured by Ni0 species 184, ultimately leading to the formation of a NiI complex 185. Subsequently, aryl halides 151 undergo oxidative addition with intermediates 185 to form key (aryl)(alkyl)NiIII species 186. This is followed by a rapid reductive elimination, which affords the final products 181 and NiI complex 187. An alternative potential pathway for the formation of intermediate 188 is the oxidative addition of Ni0184 with aryl halides 151 to give NiII species 188, which is then intercepted by radicals 183. Lastly, single-electron reduction of 187 by reducing IrII to regenerate the ground-state PC IrIII and Ni0 species 184, thereby closing two catalytic cycles.
Nickel-catalyzed reductive coupling reactions have attracted considerable attention because they rely solely on organo-electrophiles, eliminating the need for stepwise pre-synthesis of organometallic reagents. In 2020, Li et al. reported a visible-light-induced Ni-catalyzed reductive coupling of 1,3-dienes 97a with aldehydes 189, which resulted in the delivery of homoallylic alcohols 190 in yields ranging from 34% to 99% under mild conditions [Scheme 25][86]. The authors employ the Hantzsch ester (HE) as the hydrogen radical source to facilitate the oxidation of the low-valent nickel salt, thereby yielding the Ni-H species. Furthermore, this dual catalytic system is compatible with aryl or alkyl aldehydes. However, the regioselectivity for asymmetric 1,3-dienes is only 1.9:1, indicating that additional optimization is necessary to enhance the asymmetric induction.

Scheme 25. Photo- and nickel-catalyzed reductive 1,2-hydroalkyaltion of 1,3-dienes with aldehydes.
The reaction pathway begins with the formation of an excited PC *Ir under visible-light irradiation, which then undergoes reduction by iPr2NEt [Eox (iPr2NEt) = +0.65 V] via SET process to form a highly reducing IrII species and [iPr2NEt]•+, the latter accepting one electron from HE to regenerate iPr2NEt and the HE•+. The reduction potential of Ir* is E1/2red [*IrIII/IrII] = +1.21 V vs. SCE in MeCN. Meanwhile, NiII complex 191 is reduced by IrII [E1/2red (IrIII/IrII) = -1.37 V vs. SCE in MeCN], affording NiI species 192 [E1/2red (NiII/NiI) = -0.68 V vs. SCE in DMSO] and regenerating ground-state IrIII, thus closing the photocatalytic cycle. NiI species 192 then combines with HE•+ to deliver key intermediate Ni-H species 193 and pyridinium ion pyH+. The Ni-H species is captured by s-cis configuration 1,3-dienes 97a to produce the key anti-π-allyl-nickel intermediate 194, which can isomerize to syn-π-allyl-nickel intermediate 195 rapidly. Intermediates 194 and 195 are subsequently converted to Z- and E-σ-allyl nickel intermediates 196 and 197. Next, intermediates 197 react with aldehydes 189 and, through the Zimmerman-Traxler transition state 198, form syn-homoallylic nickel alkoxides 199, which are protonated by pyH+ to yield syn-homoallylic alcohols 190 and regenerate NiII complex 191 along with HP. Moreover, E-σ-allyl nickel intermediates 197 follow a comparable pathway, resulting in the formation of anti-homoallylic alcohols 190. The authors believe that the crotyl methyl group placed pseudoaxially in transition state 198 may serve to mitigate gauche interactions between this group and the substituent of the aldehydes 189, as observed in certain reactions. This may explain why transition state 198 is more favorable than 200 in this reaction. Furthermore, the latter transition state model results in anti-diastereoselectivity.
Additionally, sodium sulfinate can function as a radical precursor in the nickel-catalyzed three-component cross-coupling reaction under photochemical or electrochemical conditions. In 2020, Huang et al. reported a visible-light-induced and nickel-catalyzed cascade cross-coupling process of dienes 201 with sodium sulfinates 202 and aryl halides 151, which delivered a series of saturated five-membered ring compounds in 46%-99% yields [Scheme 26][87]. This process synthesizes three new chemical bonds (one C−S and two C−C bonds) in one step, facilitating the rapid construction of complex molecules from commercially available feedstock chemicals. Notably, this strategy is compatible with 1,3/1,4/1,5/1,6-diene, aryl halides, and sodium aryl/alkyl sulfinates.

Scheme 26. Photo- and nickel-catalyzed cyclization sulfonylarylation of 1,6-dienes.
The conversion process is initiated when photosensitizer PC is excited to highly oxidizing excited-state PC* under visible-light irradiation. Radical precursor sodium sulfinate 202 is oxidized by PC* via SET process to form sulfonyl radical 204, accompanied by the generation of photocatalyst PC-. Subsequently, the PC- reduced NiI complex 206 is reduced by another SET process, resulting in the formation of the LNi0 species 207. This process ultimately returns the system to its ground-state PC. Meanwhile, the sulfur-centered radical 204 is captured by the dienes 201 and undergoes a radical cascade cyclization reaction to furnish the cyclic radical intermediates 205, which combine with LNi0 species 207 to form alkyl-NiI intermediates 208, followed by oxidative addition with aryl halides 151 to give NiIII complex 209. Lastly, intermediate 209 undergoes reductive elimination to deliver desired product 203 and NiI species 206.
Silicon-containing compounds have a broad of applications in the fields of organic synthesis, medicinal chemistry, and material science. Zhang et al. reported a photoredox and nickel-catalyzed three-component carbosilylation of electron-deficient alkenes 150 with aryl bromides 159 and (TMS)3SiH 210 to deliver organosilicon compounds with 14%-84% yields [Scheme 27][88]. The silylation reaction involves the participation of photogenerated silicon radical intermediates, whereas the arylation process is conducted through the utilization of nickel-catalyzed cross-coupling. The arylsilylation system of alkenes is compatible with (hetero)aryl bromides, although the alkenes are limited to activated ones.

Scheme 27. Photo- and nickel-catalyzed silylarylation of electron-deficient alkenes.
The coupling conversion is carried out under visible-light irradiation with Ir(dF-Me-ppy)2(dtbbpy)(PF6) as PC and Ni(cod)2 as nickel catalyst. First, Ni0 species 212 and aryl bromides 159 undergo oxidative addition, forming NiII intermediate 213. Concurrently, the IrIII PC is excited by blue LED light, resulting in the formation of the excited-state *IrIII. This is then reductively quenched by Br- of 213, generating IrII and bromide radicals 214. Subsequently, a hydrogen atom is abstracted from (TMS)3SiH 210, producing a silyl radical 215. Following that, methyl acrylate 150 captures silyl radical 215 to generate β-silyl alkyl radicals 216, which is trapped by NiII species 213 to afford NiIII complex 217. Then, C−C reductive elimination from 217 yields the final products arylsilylation 211 and NiI species 218. Lastly, the IrII complex reduces NiI218, thereby regenerating both the Ni0 catalyst and the PC IrIII.
Notably, the addition of chiral ligands in the visible-light and nickel co-catalytic system can also result in asymmetric cross-coupling reactions of alkenes. In 2020, Guo et al. developed a visible-light-induced photoredox/nickel dual-catalyzed enantioselective three-component alkylarylation reaction of activated alkenes 150 with secondary or tertiary alkyl trifluoroborates 158 and aryl bromides 159 to deliver β-alkyl-α-arylated carbonyls, phosphonates and sulfones with 16%-92% yields and 15%-97% ee [Scheme 28][89]. The catalytic cycling mechanism follows that of Ni0/NiII/NiIII, with the stereo-determining step being the addition of radicals to tetrahedral NiII species.

Scheme 28. Photo- and nickel-catalyzed enantioselective three-component alkylarylation of alkenes.
Based on [UB3LYP-D3(BJ)/def2-SVP-CPCM(THF)] calculations, the authors put forth a proposed mechanism for this transformation. Upon irradiation, photosensitizer IrIII was excited to *IrIII, which proceeded to oxidize alkyl trifluoroborate reagent 158 to alkyl radicals 220 and IrII by single-electron oxidation. Then, alkyl radicals 220 undergo rapid and irreversible radical addition to acrylates 150, resulting in the formation of α-ester radicals 221. Meanwhile, the photosensitizer IrII facilitated the reduction of the bipyridyl-NiI222 complex to Ni0223, which was then subjected to a rapid oxidative addition with aromatic bromides 159 to provide a square planar singlet NiII species 224. Additionally, alkyl radicals 221 were observed to be captured by the 223, resulting in the formation of a NiI-alkyl species 225. This subsequently underwent an oxidative addition with aromatic bromides 159, generating a NiIII species 226. It is noteworthy that the NiIII species 226 undergoes reversible radical dissociation, forming a square planar singlet NiII species 224. This species can subsequently undergo isomerization and intersystem crossing, leading to the emergence of the more stable tetrahedral triplet spin state NiII227 species. Lastly, 227 rapidly and irreversibly combines with the α-ester radicals 221 to obtain NiIII species 226’, resulting in the formation of the alkylarylation products 219 and NiI complex 222. The authors found that the energy of the radical addition step (227 to 226’) is greater than that of the reductive elimination transition state (226’ to 222). The results demonstrate that the stereo-determining step of this process is the radical addition of the tetrahedral NiII center, rather than reduction elimination.
The poor stability of primary alkyl radicals, which are produced by photoredox/nickel dual catalysis, makes this process relatively difficult. Zheng et al. developed a photoinduced Ni-catalyzed chemo- and regioselective reductive conjugate cross-coupling reaction of alkenes 150 with aryl halides 151 and α-silyl amines 228, enabling the synthesis of α-aryl substituted γ-amino acid derivatives in 20%-90% yields

Scheme 29. Photo- and Ni-catalyzed 1,2-aminomethylarylation of electron-deficient alkenes.
The proposed mechanism is illustrated in Scheme 29. α-Silyl amines 228 are oxidized by excited-state PC* via SET route to give primary α-amino radicals 230, which rapidly undergo Giese-type addition to form
The use of unactivated C(sp3)-hybridized centers from the hydrocarbon skeleton as radical precursors will lead to a simpler photo/nickel dual-catalyzed three-component cross-coupling reaction. In 2021, Xu et al. described a visible-light-induced nickel-catalyzed three-component dicarbofunctionalization of conjugated alkenes 150 with C(sp3)-hybridized centers compounds 236 and aryl halides 151 to afford α-aryl esters in trace to 82% yields [Scheme 30][91]. The protocol employs an inexpensive and readily synthesizable decatungstate [tetrabutylammonium decatungstate (TBADT)] as a PC and abundant hydrocarbons as radical precursors, facilitating the installation of a diverse array of highly functionalized tertiary, secondary, and primary C(sp3)-hybridization centers. Furthermore, this strategy is compatible with a series of activated alkenes, aryl halides, and secondary or tertiary C−H bonds under mild conditions.

Scheme 30. Photoinduced tungsten- and Ni-catalyzed 1,2-alkylarylation of electron-deficient alkenes via aliphatic C–H cleavage.
As shown in Scheme 30, the authors proposed a possible catalytic pathway. Under visible-light irradiation, TBADT is excited to its excited-state *[W10O32]4-, which then abstracts a hydrogen atom from unactivated hydrocarbon substrates 236 to provide reduced decatungstate [W10O32]5-H+ and alkyl radicals 238. Then, methyl acrylate 150 captures alkyl radicals 238 to give another alkyl radicals 239, which are intercepted by
Similarly, Campbell et al. developed a visible-light-induced photoredox/nickel-catalyzed three-component cross-coupling of activated alkenes 150 with aryl bromides 159 and C−H bonds building blocks (alcohols, ethers lactones and lactams) 244 to afford dicarbofunctionalization products 245 in 31%-96% yields

Scheme 31. Photoinduced ketone and Ni-catalyzed 1,2-alkylarylation of electron-deficient alkenes by C–H cleavage.
Mechanism studies have shown that this process involves hydrogen bond-assisted radical addition. Firstly, upon irradiation, diaryl ketone 246 is excited to triplet-state diradical 247, which interacts with 244 to afford carbon-centered radicals 248 and ketyl radicals 249 through selective homolysis of an activated C−H bond. Subsequently, the alkyl radicals 248 undergo a regioselective Giese addition reaction with activated alkenes 150, forming the radical adducts 250. These are captured by a Ni0 species 251, resulting in the formation of the NiI complex 252. Finally, oxidative addition with aryl bromides 159 occurs, leading to the generation of the NiIII intermediate 253. Conversely, Ni0 may also undergo oxidative addition directly with aryl bromides 159, yielding tetrahedral NiII intermediates 254. These could then combine with radicals 250 to form the NiIII intermediates 253. Next, reductive elimination from 253 would produce the final products 245 and NiI species 255. Ultimately, the ketyl radical 249 reduces the NiI species 255, thereby regenerating the Ni0 species 251 and returning the ground-state diaryl ketone 246 to its original state through SET process.
Jiang et al. reported a photoredox and nickel-catalyzed three-component aminoarylation of electron-rich alkenes 150 with 2,2,2-trifluoroethoxy carbonyl protected α-amino-oxy acids 256 and aryl bromides 159, which provide 1,2-aminoarylation products 257 in 37%-89% yields [Scheme 32][93]. The anti-Markovnikov addition of the amidyl radicals derived from 256 to the alkenes 150 and Ni-mediated radical/transition metal crossover result in the formation of 1,2-aminoarylation product. The category of electron-rich alkenes encompasses a number of different compounds, including vinyl ethers, vinyl amides, vinyl amines and vinyl thioethers.

Scheme 32. Photo- and Ni-catalyzed 1,2-aminoarylation of electron-rich alkenes.
The catalytic process commences with the photoexcitation of 4CzIPN upon irradiation to generate the excited redox catalyst 4CzIPN*, which oxidizes deprotonated 256 by SET pathway, resulting in the formation of carboxyl radicals 258 and reduced 4CzIPN•-. The cleavage of radicals 258 results in the formation of nitrogen-centered radicals 259, accompanied by the release of CO2 and acetone. Subsequently, electron-rich alkenes 150 are captured by radicals 259 via radical addition, affording the radical adducts 260. Concurrently, the reduced photosensitizer 4CzIPN•- reduces NiI species to Ni0 and returns to the ground-state 4CzIPN, closing the photocatalytic cycle. Next, Ni0 undergoes oxidative addition with aryl bromide 159 and combines with radical adducts 260 to form key NiIII complex 262, which is then subjected to reductive elimination to afford final products 257 and a NiI species, completing the nickel catalysis cycle.
Enantioenriched α-aryl propionic acid is an important structural unit of nonsteroidal anti-inflammatory medications (NSAIDs). Qian et al. reported a visible-light-promoted photoredox/nickel dual catalyzed three-component reductive coupling reaction of acrylates 150 with alkyl halides 175 and aryl bromides 159, which afforded enantioenriched α-arylpropionate esters 263 with 15%-90% yields and 33%-93% ee

Scheme 33. Photo- and Ni-catalyzed enantioselective 1,2-alkylarylation of acrylates under reductive conditions.
The plausible mechanism of this reaction is proposed in Scheme 33. The reduced PC 4CzIPN•- via two SET processes in situ provides LNi0 species 264, which undergoes oxidative addition with the aryl bromides 159 to yield the LNi(Ar)IIBr complex 265. Meanwhile, the alkyl bromides 175 undergo a SET process by either the 264 or reduced 4CzIPN•- to generate alkyl halide radical anions, which then undergo a loss of bromide, generating the tertiary radicals 267, being tapped by acrylate 150 and deliver α-carbonyl radical 268. Subsequently, radical 268 is intercepted by 265 to form NiIII complex 269, which undergoes rapid reductive elimination to give the final products 263 and NiI species 266. In the photoredox cycle, photoexcited
In the reported visible-light-mediated/nickel dual-catalyzed alkenes three-component cross-coupling reactions, many catalytic systems require the introduction of PCs to initiate the reaction. Interestingly, the difunctionalization of alkenes can also be achieved without the use of external PCs through the ingenious design of substrates. In 2022, Xi et al. developed a visible-light-induced nickel-catalyzed three-component net-reduction 1,2-alkylacylation of activated alkenes 150 with NHPI esters 113 and 2-pyridyl esters 271 to furnish a series 1,3-dicarbonyl compounds 272 with 30%-81% yields [Scheme 34][95]. Notably, the reductive coupling generates electron donor-acceptor (EDA) complexes in situ, obviating the need for the addition of external PCs and stoichiometric metal reducing agents. Additionally, this reaction is applicable to a range of activated alkenes, NHPI esters, and 2-pyridine esters under mild conditions.

Scheme 34. Photo- and Ni-catalyzed 1,2-alkylacylation of electron-deficient alkenes.
The mechanism study shows that the reaction begins with the coordination of NHPI esters 113 and HE to form photoactive EDA complex 273 under visible-light irradiation, which results in the extrusion of CO2 and phthalimide anion and subsequently generates alkyl radical 274. The latter is then captured by activated alkenes 150 through Giese addition, leading to the formation of radical intermediates 275. Meanwhile, the catalytically active Ni0 is formed by the reduction of NiII pre-catalyst 276 with the excited state of HE*. Then, Ni0 species combine with alkyl radical 275 to provide the NiI complex 277, which undergoes oxidative addition with the ester 271 leading to the generation of NiIII intermediate 278. An alternative pathway involves the direct oxidative addition of Ni0 to 2-pyridyl esters 271, resulting in the formation of NiII complex 279. This complex then interacts with alkyl radicals 275, generating NiIII intermediates 278. Finally, species 278 undergo reductive elimination to furnish the 1,3-dicarbonyl compounds 272 and NiI species 276. Lastly, the excited state of HE* reduces 276, thereby regenerating active Ni0 species and closing the catalytic cycle.
In general, the regioselectivity of electronically unbiased alkyl alkenes is challenging to control. However, this can be addressed by introducing directing groups into the raw materials, which allows for greater control over the reaction outcome. Dey et al. reported a visible-light-mediated and nickel dual catalyzed three-component 1,2-dicarbofunctionalization of unactivated alkenes 280 with alkyl bromides/iodides 25 and aryl iodides 168 to deliver coupling products 281 in 32%-84% yields [Scheme 35][96]. Notably,

Scheme 35. Photo- and Ni-catalyzed 1,2-arylalkylation of unactivated alkenes.
The transformation is initiated by visible-light. Upon irradiation, NiII from NiBr2 is reduced to NiI species by PC IrIII, which coordinates with 8-aminoquinoline and then tethers unactivated alkenes 280 to generate NiI complex 282, followed by sequential oxidative addition and migration insertion providing NiIII complex 283. Next, the reduced-state IrII undergoes SET process to reduce 283 to NiII species 284, accompanied by the generation of ground-state IrIII. Meanwhile, tertiary amine 285 or HE reacts with *IrIII via a SET pathway to obtain α-amino radical cations 286 or HE•+, which undergo a rapid deprotonation process leading to the generation of α-amino radicals 287. Subsequently, they interact with alkyl halides 25 through halogen atom transfer (XAT), producing alkyl radicals 288. Lastly, alkyl radicals 288 are trapped by intermediates 284 to give NiIII species 289, which then undergo reductive elimination to deliver final products 281.
The exceptionally high reactivity of open-shell radical species has resulted in the catalytic asymmetric multicomponent cross-coupling involving radicals remaining largely underdeveloped. In 2023, Li et al. reported a nickel metallaphotoredox catalyzed chemo-, regio-, stereo-, and enantioselective three-component cross-coupling reaction of vinyl phosphonates 290 with alkenyl halides 291 and alkyl trifluoroborates 158 to produce a series of enantioenriched α-alkenyl phosphonates 292 with 43%-89% yields and 85%-98% ee [Scheme 36][97]. Moreover, by modifying the triplet energy of the PC, it is possible to achieve the divergent formation of enantio-enriched trans and cis α-alkenyl phosphonates. The strategy is compatible with various alkenyl halides 291 and alkyl trifluoroborates 158 under mild conditions, with the exception that the alkene contains a vinyl phosphate ester.

Scheme 36. Photo- and Ni-catalyzed enantioselective 1,2-alkylalkenylation of vinylphosphonates.
The possible mechanism of this three-component reaction is shown in Scheme 36. Upon irradiation, ground-state PC IrIII is converted into its excited-state *IrIII, which oxidizes alkyl trifluoroborates 158 to alkyl radicals 293 by single-electron oxidation. This is followed by a fast and irreversible Giese addition to vinyl phosphonates 290, which affords α-phosphonate alkyl radicals 294. Simultaneously, generated Ir- reduces NiI complex 295, which is derived from nickel coordinating with chiral ligands to the Ni0 species 296 along with the regeneration of the ground-state IrIII and the termination of the photocatalytic cycle. Then, Ni0 species 296 engage with vinyl bromides 291, resulting in direct oxidative addition to generate singlet spin-state, square-planar NiII297. This then undergoes isomerization to the tetrahedral, triplet spin-state NiII298, followed by the capture of α-phosphonate alkyl radicals 294, which affords the crucial NiIII species 299. An alternative pathway involves the direct combination of Ni0 with alkyl radicals 294 via radical addition, resulting in the formation of NiI complex 300. This complex then undergoes oxidative addition with vinyl bromides 291, leading to the generation of NiIII species 299. Finally, NiIII species 299 undergoes reversible radical dissociation prior to productive reductive elimination to give the desired products 292 and regenerate NiI complex 295, thereby closing the nickel catalytic cycle.
At present, there are still rare reports on the three-component cross-coupling reaction of aromatic alkenes that employ a combination of photoredox and nickel catalysis. In 2023, Liu et al. developed a visible-light-induced and nickel-catalyzed three-component enantioselective transformation of aryl alkenes 12 with alkenyl halides 291 and sodium sulfinates 202 to afford enantioenriched β-chiral sulfones 301 in 45%-90% yields and 72%-94% ee [Scheme 37(a)][98]. This dual catalytic strategy allows for a one-step reaction in the presence of chiral ligands while controlling enantioselectivity, and thus enables the direct obtaining of the target product from readily available starting materials. Moreover, this protocol is applicable to a range of alkenyl halides and sodium aryl/alkyl sulfinates, although the alkenes are limited to terminal aromatic alkenes.

Scheme 37. Ni-organophotocatalyzed enantioselective sulfonylalkenylation of styrenes.
This process is initiated by the PC Eosin Y being excited to excited-state Eosin Y* under visible-light irradiation. This results in the oxidation of sulfinates 202, which deliver the radical anion Eosin Y•- and sulfonyl radicals 302. The latter is trapped by styrenes 12, leading to the formation of benzylic radicals 303. Meanwhile, active species Ni0304 reacts with alkenyl halides 291 by oxidative addition to form alkenyl nickel intermediates 305. These selectively rebond with benzylic radicals 303 to form L*NiIII complex 306, which then undergoes reductive elimination to provide the final β-chiral alkenyl sulfones 301 and L*NiIX species 307. Lastly, L*NiIX is reduced by Eosin Y•- to regenerate L*Ni0 species 304 and ground-state Eosin Y, and accompanied by the close of two catalytic cycles.
Similarly, Du et al. described a nickel/photoredox dual-catalyzed asymmetric three-component carbonsulfonylation of N-vinylamide 150 with (hetero)aryl halides 151 and sodium sulfinates 202[99]. This process simultaneously forms C−C and C−S bonds, allowing the synthesis of β-aryl and β-alkenyl sulfones in 40%-92% yields and 88:12-99:1 er [Scheme 37(b)]. The strategy employed 4CzIPN as the PC, chiral biimidazoline as the ligand, and is completed through Ni0/NiI/NiIII work pathway. Differently, secondary alkyl radicals 303 are rapidly captured by L*Ni0 complex 304 to afford (alkyl)NiI species. Next, aryl halides 151 and (alkyl)NiI species undergo oxidative addition to give (aryl)(alkyl)NiIII intermediates 306.
In accordance with the general mechanism proposed in Scheme 19, radical precursors are typically assembled at the distal carbon of alkene, while aromatic groups are located at the proximal carbon. Consequently, the alternative assembly mode, in which the radical precursor is attached to the proximal carbon of the olefin and the aryl group is attached to the distal carbon of the alkene, can facilitate the formation of more flexible and diverse coupling products. In 2023, Zhao et al. developed a nickel/photoredox dual catalyzed three-component reductive cross-coupling of electron-deficient alkenes 150 with aryl iodides 168 and aldehydes 189 [Scheme 38][100]. This process afforded β-hydroxy ester/amide compounds 308 with 30%-81% yields and 1:1-1:1.8 dr. Notably, the success of this coupling/tandem transformation hinges on the identification of α-silylamine as the organic reductant. This enables the release of silylium ions in lieu of protons, thereby preventing any unwanted protonation processes. Additionally, it serves as a Lewis acid to facilitate the in situ activation of aldehydes. The method is applicable to a range of substrates, including acrylic esters and amides, along with alkyl and aryl aldehydes.

Scheme 38. Photo- and Ni-catalyzed reductive arylalkylation of alkenes with aldehydes.
A possible reaction mechanism is shown in Scheme 38. Initially, two SET processes occur between the photoexcited *IrIII (EIV/*III1/2 = -1.73 V vs. SCE) and NiII [E1/2(NiII/Ni0) = -1.2 V vs. SCE] resulting in the generation of IrIV and LNi0 species. The latter exhibits strong reductive potential IrIV (EIV/III1/2 = +0.77 V vs. SCE), which can oxidize α-silylamine (Eox1/2 = +0.71 V vs. SCE) to get iminium ion and TMS+ through another two SET processes, along with ground-state IrIII. Concurrently, Ni0 species 309 undergoes oxidative addition with aryl iodides 168 to afford NiII species 310, of which the migration insertion into C=C bond of 150 occurred to give α-carbonyl-NiII complex 311. An equilibrium exists between NiII oxygen enolate 312 and carbon enolate 311 tautomers, which could be further transformed into a more stable silyl ketene acetal 313 via a Ni/Si transmetallation. Lastly, the aldol reaction, which is promoted by Lewis acidic silylium ions, delivers intermediates and regenerates the NiII catalyst. The removal of TMS, performed using tetrabutylammonium fluoride (TBAF), provides the final products.
In 2024, Ye et al. developed a visible-light-induced photoredox/nickel dual catalyzed ligand-controled regioreversed 1,2-arylalkylation of activated alkenes 150 with aryl halides 151 and α-silylamines 228[101]. This process furnished β-amino esters/amides 316 in 22%-92% yields [Scheme 39]. Notably, this strategy involves the generation of phenyl radicals for the attachment of the phenyl group on the distal carbon of alkene, rather than the conventional Giese addition. Through mechanism studies, the authors identified ortho-substituted bipyridine ligands as the key factor in regulating regioselectivity. In addition, this strategy is suitable for both aryl halides and vinyl halides under mild conditions, although it is not compatible with primary or secondary amines.

Scheme 39. Photo- and Ni-catalyzed alkylaminoarylation of electron-deficient alkenes.
The authors put forth two possible reaction pathways. In cycle A, the excited photosensitizer PC* reacts with α-silylamines 228 by single electron reduction to form α-amino radicals 317. These radicals are then trapped by alkenes 150 by Giese addition to form α-carbonyl radicals 318, followed by combining with Ni0 complex 319 to give NiI intermediates 320. Subsequently, oxidative addition of 320 with aryl halides 151 results in the formation of NiIII intermediates 321, which undergo reductive elimination to yield the 1,2-aminoalkylarylation products 316 and NiI complex 322. Finally, the SET process between NiI322 and the reduced state PC•- to regenerate ground-state PC and 319. Notably, the use of bpy ligands with strong coordination ability inhibits the competitive XAT pathway for the reaction of aryl halides with Ni0 complexes. Furthermore, the ortho-substituted bpy ligand weakens the coordination affinity of alkenes to the Ni0 center, thus enhancing the reactivity of XAT to generate aryl radicals 323, which in turn trigger the 1,2-arylaminealkylation process (cycle B). Therefore, the reaction is preferentially initiated by the metal-alkene coupling radical addition reaction of 323, forming triplet alkyl NiII324. This then undergoes homolytic substitution with α-amino radicals 317, resulting in the formation of the 1,2-arylaminoalkylation products 316.
The chlorine radical is widely acknowledged as an effective electrophilic hydrogen atom abstractor. In 2024, Koo et al. reported a nickel/photoredox-catalyzed three-component silylacylation of acrylates 150 with hydrosilyl compounds 325 and aryl acyl chlorides 326, which provided 1,3-dicarbonyl compounds 327 in 40%-79% yields under redox-neutral conditions [Scheme 40][102]. The protocol employs LMCT to facilitate the cleavage of NiIII-Cl, thereby generating chlorine radicals. These radicals then activate Si−H bonds through hydrogen atom transfer (HAT), resulting in the generation of silicon radicals. Notably, acyl chlorides serve the dual function of acylating agents and chloride donors throughout the process. Additionally, this catalytic system is suitable for the conversion of acrylic esters and amides, but only compatible with arylacyl chlorides and tertiary silanes.

Scheme 40. Photo- and Ni-catalyzed silylacylation of acrylates.
The reaction is initiated by the oxidative addition reaction of LNi0328 with aryl acyl chlorides 326 to form NiII329, which undergoes single-electron oxidation by the excited PC *IrIII (*E1/2 = +1.21 V vs. SCE in MeCN), leading to the formation of NiIII species 330. Upon visible-light irradiation, the NiIII complex 330 engages in LMCT, resulting in the photoelimination of a chlorine radical 331 and the regeneration of a NiII complex 332. This chlorine radical then interacts with hydrosilanes 325 by HAT, affording silyl radicals 333. These are subsequently captured by α,β-unsaturated alkenes 150 to give alkyl radicals 334, followed by combining with 332 to yield NiIII species 335. This then undergoes reductive elimination to produce the 1,3-dicarbonyl compounds 327 and a NiI complex 336. Finally, the reduction of IrII (E1/2 = -1.37 V vs. SCE in MeCN) by NiI (Ep = -1.17 V vs. SCE in THF) via SET results in the regeneration of ground-state IrIII and the formation of Ni0 species 328, thereby completing the dual catalytic cycle.
The construction of C(sp3)−C(sp3) bonds represents a significant challenge in cross-coupling chemistry. In 2024, Cong et al. reported a photoredox/nickel dual catalyzed trimolecular cross-coupling of unactivated alkenes 8 with alkyl halides 25 and hypervalent iodine-based reagents 337 to deliver dicarbofunctional products 338 in 29%-94% yields [Scheme 41][103]. Notably, the approach undergoes a bimolecular homolytic substitution (SH2) mechanism and chemoselective XAT to achieve regioselectivity without the assistance of a directing group and simultaneously constructing two C(sp3)−C(sp3) bonds.

Scheme 41. Photo- and Ni-catalyzed dialkylation of unactivated alkenes.
Irradiation of the PC 4CzIPN with blue light resulted in the conversion of 4CzIPN into a high-energy triplet state 4CzIPN*. This energy transfer to the iodine (III) compound 337 was facilitated by triplet-triplet energy transfer (TTenT), leading to the homolysis of the I−O bond and the subsequent formation of carboxyl radicals. These radicals fragment to release methyl radical 339, iodobenzene, and CO2. Subsequently, the nucleophilic and unhindered 339 is trapped by the in situ-generated NiII species 340 to form the NiIII species 341. Meanwhile, another equivalent of methyl radical engages with the electron-poor alkyl iodides 25 by chemoselective XAT to afford an electrophilic alkyl radical 342 and an iodomethane byproduct. Alkyl radicals 342 are regioselectively intercepted by the unactivated C=C bond of 8 (polarity matching) to form a sterically more congested alkyl radical 343, which favors SH2 coupling with 341 to afford NiIII complex 344. This complex is then delivered to final dialkylation products 338 and regenerate 340, thereby shutting off the nickel catalytic cycle. It is crucial to note that the success of this dialkylation process depends on the ability of the organonickel-catalyzed species to discern between various radical intermediates 339, 342 and 343 (the nucleophilic and unhindered 339 preferentially associates with the Ni center in 340), and the key role of the electrophilic reagents 342 is to facilitate the nucleophile 339 induced selective XAT formation reaction with 25.
Wang et al. reported a visible-light-induced nickel-catalyzed three-component arylalkylation of unactivated alkenes 8 with aryl bromides 159 and NHPI esters 113 to afford desired products 345 in 41%-83% yields

Scheme 42. Photo- and Ni-catalyzed arylmethylation of alkenes.
The process starts with the iridium PC IrIII being excited to long-lived triplet excited-state
In 2024, Hu et al. described a visible-light-promoted and nickel-catalyzed asymmetric three-component dicarbofunctionalization of alkene 150 with C(sp3)–H nucleophiles 236 and aryl bromides 159 to provide

Scheme 43. Photo- and Ni-catalyzed enantioselective alkylarylation of alkenes.
The catalytic mechanism is illustrated in Scheme 43. First, ground-state PC [W10O32]4- is stimulated to excited-state *[W10O32]4- by visible-light, which then abstracts a hydrogen atom from C(sp3)-H nucleophiles 236, resulting in the formation of alkyl radicals 352 and singly reduced [W10O32]5-H+. The disproportionation of [W10O32]5-H+ results in the formation of doubly reduced [W10O32]6-2H+ and the regeneration of ground-state [W10O32]4-. The photocatalytic cycle was completed. Subsequently, carbon-centered radicals 352 are intercepted by alkenes 150 via radical addition to another alkyl radical 353. The latter species is then trapped by Ni0 species 354, resulting in the formation of alkyl-NiI species 355, which undergoes oxidative addition with alkenyl bromides 159 to generate alkyl-NiIII-aryl complex 356. Alternatively, NiI species 357 could combine with alkyl radicals 353 to form alkyl-NiII species 358, which are converted to alkyl-NiI species 355 via single-electron reduction. Finally, intermediates 356 undergo reductive elimination to produce the final products 351 and NiI species 357, which interact with doubly reduced [W10O32]6-2H+ via a SET route, thereby regenerating the active Ni0 catalyst 354 and singly reduced [W10O32]5-H+.
PHOTOREDOX AND CHROMIUM-CATALYZED THREE-COMPONENT DIFUNCTIONALIZATIONS OF ALKENES
In recent years, chromium catalysis has attracted considerable interest due to its distinctive reactivity[13]. However, three-component cross-coupling reaction of alkenes catalyzed by the combination of chromium and visible-light remains underdeveloped.
In 2020, Schwarz et al. developed a photoredox and chromium-catalyzed regioselective and diastereoselective dialkylation of 1,3-dienes 97a with aldehydes 189 and 4-alkyl HEs 359 to deliver homoallylic alcohols 360 in 51%-95% yields and 70:30 to > 95:5 dr [Scheme 44][106]. Moreover, enantioselective synthesis of homoallylic alcohols can be achieved by incorporating chiral bisoxazoline ligands into the catalytic system. This approach is applicable to diverse alkyl/aryl aldehydes, 4-alkyl HEs and alkyl-substituted 1,3-dienes.

Scheme 44. Photo- and Cr-catalyzed 1,2-dialkylation of 1,3-dienes.
Mechanistic studies indicate that the coupling reaction begins with the excitation of PC to its highly oxidizing excited-state PC*, which is quenched by 4-alkyl HEs 359 to generate alkyl radicals 361 and pyridinium species pyH+, along with the reduced photocatalyst PC•-. Subsequently, the alkyl radicals 361 could either reversibly interact with low-valent LCrII species 362 to give an off-cycle LCrIII species 363 or react with 1,3-dienes 97a to give allyl radicals 364. The allyl radicals 364 were captured by LCrII species 362 to afford the CrIII allyl species 365 and 366. Notably, the formation of chromium alkoxide 367 via a six-membered Zimmerman-Traxler transition state would occur in a regioselective and anti-selective manner. Subsequently, hydrolysis of 367 by pyH+ gives the formation of products 360 and LCrIII complex 368. A SET process between the PC•- and 368 regenerates the PC and LCrII362.
In 2022, Liu et al. reported a flexible visible-light-mediated chromium-catalyzed radical diacylation of alkenes 12 or 1,3-dienes with α-keto acids 369, which afforded dione compounds 370 in 29%-81% yields

Scheme 45. Photo- and Cr-catalyzed diacylation of alkenes.
The plausible mechanism of this reaction is also illustrated [Scheme 45]. First, PC IrIII is transformed to its excited state *IrIII under visible-light irradiation. Meanwhile, base-mediated deprotonation of phenylglyoxylic acid 369 results in the formation of α-oxocarboxylate 371, which is then oxidized to acyl radicals 372 by *IrIII by releasing CO2 as a byproduct. Alternatively, arylglyoxylic acids 369 could be responsible for producing acyl radicals 372 and carboxyl radicals 373. Carboxyl radicals 373 can oxidize IrII PC back to IrIII, thus completing the photocatalytic cycle. On the other hand, acyl radicals 372 are intercepted by aryl alkenes 12 to yield benzyl radicals 374. In parallel, anionic CrI species may be produced via the single-electron reduction of CrII catalyst by PC IrII, which combine with acyl radicals 372 to form CrII complex 375 and further trapped benzyl radicals 374 to form stable quartet CrIII intermediates 376. Finally, CrIII intermediates 376 undergo reductive elimination to form the final 1,4-dione products 370 and a CrI species 377.
In 2024, Wu et al. developed a visible-light and chromium-catalyzed regio- and diastereoselective radical three-component cross-coupling reaction of 1,3-enynes 97b with alkyl halides 25 and aldehydes 189 to provide a series of homopropargylic alcohols 378 in 43%-85% yields with 3:1 to > 20:1 dr [Scheme 46][108]. Importantly, the dialkylation of 1,3-enynes proceeds in the absence of external PCs, stoichiometric metal reductants, and additives. This protocol could be applied to 1,3-enynes and alkyl electrophilic reagents.

Scheme 46. Photo- and Cr-catalyzed reductive 1,2-dialkylation of 1,3-enynes.
The possible mechanism of the three-component reaction involving 1,3-enyne is illustrated in Scheme 46. Initially, upon irradiation with blue light, HE is transformed into its active state HE*, which then reduces alkyl halides 25 by a SET process to form the alkyl radicals 379 and HE•+. Akyl radicals are further captured by C=C bonds of 1,3-enynes 97b to give the propargyl radicals 380. Meanwhile, HE•+ loses an electron and a proton, resulting in the formation of the pyridinium pyH+. Next, the propargyl radicals 380 are intercepted by CrII species to yield the nucleophilic propargylic CrIII complex 381. This complex is in equilibrium with the resonance form of the allenic CrIII complex 382, which thereafter interacts with aldehydes 189 by nucleophilic addition, leading to the generation of the intermediate 383 with high diastereoselectivity via an anti-transition six-membered cycle. Subsequently, the homopropargylic alcohols 378 and CrIII species are formed by the hydrolysis of the O–CrIII bond in 383 by the pyH+. Finally, the excited HE* reduces CrIII to CrII, thereby completing the catalytic cycle.
In 2024, Hu et al. described a dual visible-light-induced chromium catalyzed three-component Nozaki-Hiyama-Kishi type reaction of 1,3-butadienes 97a, 1,3-dioxolane 384, and aldehydes 189 to afford homoallylic alcohols 385 in yields ranging from 29% to 84% [Scheme 47][109]. Notably, the photocatalyst diaryl ketone serves two distinct roles: first, in the HAT process, and second, in facilitating the turnover of the chromium catalyst. Additionally, aryl/alkyl aldehydes are suitable substrates for combination with

Scheme 47. Photo- and Cr-catalyzed 1,2-dialkylation 1,3-dienes enabled by hydrogen atom transfer.
Mechanistic studies have confirmed that the reaction is initiated by the excitation of photocatalyst diaryl ketone 386 to oxygen radical 387, which results in the abstraction of a hydrogen atom from 1,3-dioxolane 384 to give radical intermediate 388. The generation of ketyl radical 389 is a concomitant effect of this reaction. Then, the addition of 388 to 1,3-butadienes 97a affords allyl radicals 390, which interact with LCrII species 391 to form π-allyl chromium species 392. This is followed by a nucleophilic addition to aldehydes 189, forming CrIII species 393. Finally, final products 385 and LCrIII394 are obtained through the protonation reaction of free protons or protonated diaryl ketones. The reduction reaction between ketyl radicals 389 and 394 results in the regeneration of ground-state diaryl ketone 386 and CrII species 391, thereby closing two catalytic cycles.
Interestingly, chromium can also serve as a catalyst for the three-component reaction of alkenes in the presence of other earth-abundant metals under light irradiation. In 2024, Yan et al. developed a photoredox chromium and cobalt dual catalyzed allylation reaction of 1,3-butadienes 97a with aldehydes 189, which resulted in the delivery of homoallylic alcohols 395 with 45%-95% yields and 3:1 to > 20:1 dr

Scheme 48. Photo- and Cr-catalyzed hydroalkylation of 1,3-dienes with aldehydes.
As shown in Scheme 48, a possible catalytic cycle is proposed. Upon visible-light irradiation, 4CzIPN is excited to 4CzIPN*, which interacts with HE by SET to form 4CzIPN•- and HE•+; the latter continues to participate in the reaction by losing electrons and protons. The reduction of CoII and CrIII to CoI and CrII, respectively, by 4CzIPN•- is followed by the capture of a proton by CoI, leading to the formation of the
PHOTOREDOX AND TITANIUM-CATALYZED THREE-COMPONENT DIFUNCTIONALIZATIONS OF ALKENES
In 2021, Li et al. reported a visible-light-mediated titanium-catalyzed three-component reaction of

Scheme 49. Photo- and Ti-catalyzed of 1,2-dialkylation of 1,3-dienes.
Under visible-light irradiation, the ground-state PC IrIII is excited to excited-state *IrIII to initiate the catalytic cycle. *IrIII can reduce alkyl halides 175 via a SET pathway, forming alkyl radicals. These radicals are then rapidly trapped by 1,3-dienes 97a, generating the allyl radicals 402. Then, allyl radicals 402 combine with TiIII to afford key intermediate π-allyltitanium complexes 403, which undergo nucleophilic addition with aldehydes or ketones 189 to afford TiIV species 404. Subsequently, following hydrolysis by pyH+, the homoallylic alcohols 401 are formed and release a free TiIV; the latter is reduced to TiIII by another *IrIII through SET process and accompanied by the formation of strong oxidant IrIV. Lastly, the IrIV species is reduced by HE to ground-state IrIII, producing HE•+. This can further participate in electron transfer events, ultimately leading to the production of pyH+. Notably, the anti-selectivity of homoallylic alcohols can be explained by Zimmerman-Traxler transition state.
The next reaction involved the use of thioethers or thiols in place of alkyl halides, leading to the discovery of a three-component regio- and diastereoselectivity process for 1,3-butadiene 97a with thioethers/thiols 400 and aldehydes 189 under visible-light and titanium catalysis. This process resulted in the formation of
Yan et al. developed a photoredox CoPor and Cp2TiCl2 dual catalyzed radical reaction of 1,3-butadienes 97a with aldehydes 189, which afforded homoallylic alcohols 405 with 38%-92% yields and 9:1 to > 20:1 dr

Scheme 50. Photo- and Ti-catalyzed crotylation of aldehydes with 1,3-butadiene.
The plausible mechanism of the above transformation is shown in Scheme 50. First, the photoexcited
PHOTOREDOX AND MANGANESE-CATALYZED THREE-COMPONENT DIFUNCTIONALIZATIONS OF ALKENES
In 2022, the significant reactivity of cytochrome P450 hydroxylases and non-heme iron-dependent oxygenases inspired further research. Bian et al. first developed visible-light-induced and manganese-catalyzed difunctionalization of unactivated alkyl alkenes 8 with fluoroalkylation reagents 16 and anionic nucleophiles 409 by ATRA-diverting radical ligand transfer (RLT), affording difunctionalization products 410 in 42%-92% yields [Scheme 51][115]. The nucleophiles, including TBA-Cl, TMS-N3, TBA-SCN and TMS-NCS, realized chloro-, azido-, and thiocyanato(fluoro)alkylation of unactivated alkenes. Additionally, this RLT elementary step involves a coordinated nucleophile rebounding to a carbon-centered radical, forming a new C−X bond in analogy to the radical rebound step observed in metalloenzymes.

Scheme 51. Photo- and Mn-catalyzed alkylchorination of unactivated alkenes.
In 2023, Li et al. reported a visible-light-promoted manganese-catalyzed hydrosulfonylation of alkenes 8 with sulfonyl chlorides 415 and (TMS)3Si-H 210, which provides sulfonyl-containing compounds in 35%-97% yields without external PC [Scheme 52][116]. Compared to the existing photocatalytic alkenes sulfonylation methods, this technique avoids the utilization of toxic 4-hydroxythiophenol, thereby offering a green and sustainable alternative method for alkene sulfonylation. Moreover, this approach exhibits a broad substrate scope, encompassing a range of alkyl/aryl alkenes and alkyl/aryl sulfonyl chlorides.

Scheme 52. Photo- and Mn-catalyzed hydrosulfonylation of alkenes.
As shown in Scheme 52, the authors proposed a possible mechanism for this conversion. The Mn−Mn bond in L2Mn2(CO)6 is cleaved under 6 W blue LEDs, resulting in the formation of LMn(CO)3•. This subsequently abstracts a Cl atom from sulfonyl chloride 415, leading to the generation of the sulfonyl radical 417 and the production of LMn(CO)3Cl. Then, sulfonyl radicals 417 undergo radical addition, which is trapped by alkenes 8 to yield the carbon-centered radicals 418. These then abstract the hydrogen atom from (Me3Si)3SiH 210, resulting in the formation of products 416 and the silyl radicals 419. Radical LMn(CO)3• regeneration occurs via two distinct pathways. Firstly, 419 abstracts the Cl atom from LMn(CO)3Cl thereby providing (Me3Si)3SiCl as well as regenerating LMn(CO)3• (path a). Secondly, the transfer of the Cl atom from sulfonyl chloride 415 to 419 affords (Me3Si)3SiCl and regenerates 417 (path b).
PHOTOREDOX AND IRON-CATALYZED THREE-COMPONENT DIFUNCTIONALIZATIONS OF ALKENES
To date, there are few reports on the three-component cross-coupling reaction of alkenes by merging photoredox and iron catalysis. In 2024, Tang et al. described a photoredox and iron-catalyzed three-component 1,2-thiosulfonylation of aryl alkenes 8 with thiophenols 420 and sulfonyl chlorides 415, affording β-sulfur substituted sulfone compounds 421 in 40%-85% yields [Scheme 53][117]. This strategy uses readily available sulfonyl chlorides as the sulfonation reagent, enabling the simultaneous construction of two C(sp3)–S bonds, avoiding the pre-synthesis of thiosulfates. Moreover, this method is applicable to a wide range of aryl/alkyl alkenes, aryl/alkyl sulfonyl chlorides, and arylthiophenols.

Scheme 53. Photo- and Fe-catalyzed 1,2-thiosulfonylation of alkenes.
The catalytic process of this reaction is proposed. First, PC is stimulated to its excited state PC*, which reacts with sulfonyl chlorides 415 to give the sulfonyl radicals 417 and a PC•+. Then, the resulting sulfonyl radicals 417 add to alkenes 8 by radical addition, thereby generating more stable benzyl radicals 422. Simultaneously, the thiophenols 420 interact with FeII catalyst to form an iron-sulfur intermediate 423. This intermediate subsequently rebounds with the benzyl radicals 422, leading to the generation of FeIII species 424. Lastly, intermediate 424 undergoes reductive elimination and subsequent SET to PC•+, delivering the final products 421 and regenerating FeII catalyst to close the catalytic cycle.
CONCLUSION AND OUTLOOK
Over the past decades, visible-light and earth-abundant-metal enabled synergistic catalytic three-component cross-coupling reaction involving alkenes has emerged as a platform for the development of novel and highly enabling synthetic methodologies, enabling the advancement of chemical transformations that are difficult to achieve with a single catalytic cycle. Multiple chemical bonds, including carbon-carbon and carbon-heteroatom bonds, are forged in one step under mild conditions to provide rapid access to molecular complexity in a selective manner. However, research in the three-component reaction area remains quite limited. First, the combination of visible-light with copper and nickel is more investigated, yet the combination of visible-light with other earth-abundant transition metals, such as scandium, vanadium, cobalt, iron, chromium, and manganese, has received less attention. Second, the reaction mode is limited to the interaction with alkenes to form more stabilized alkyl radicals, which are then trapped by the transition metal to catalyze the bond-forming process. Moreover, only carbon-carbon bond formations are intensively investigated in the transition-metal-catalyzed step. Third, most three-component reactions are limited to biased alkenes or alkenes with a directing group. Aliphatic alkenes without electronic bias or directing groups present ongoing challenges regarding reactivity and selectivity control. Fourth, catalytic enantioselective transformations using three-component reactions from alkenes remain underexplored. Most future efforts should focus on solving these limitations, enabling the forging of diverse carbon-carbon and carbon-heteroatom bonds. We hope the visible-light mediated earth-abundant transition-metal catalyzed three-component reaction of alkenes will evolve into a powerful and adaptive tool for the constructing of molecular complexity from readily available starting materials with the control of chemo-, regio-, and enantioselectivities.
DECLARATIONS
Acknowledgments
We sincerely thank all the leading chemists and co-workers involved in the development of visible-light and earth-abundant-metal-catalyzed three-component reactions of alkenes.
Authors’ contributions
Writing the manuscript: Jia, J.S.
Revising the manuscript: Chen, X.Y.; Li, Y.L.; Shu, W.
Guiding the work: Li, Y.L.; Shu, W.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (22171127, 22371115, 22373056), Sichuan Science and Technology Program (2024ZYD0017), Natural Science Foundation of Sichuan Province (2025ZNSFSC0128), The Pearl River Talent Recruitment Program (2019QN01Y261), Shenzhen Science and Technology Innovation Committee (JCYJ20240813094226034, JCYJ20220530114606013, JCYJ20230807093522044), Guangdong Provincial Key Laboratory of Catalysis (2020B121201002), Scientific Research and Innovation Team Program of Sichuan University of Science and Engineering (No. SUSE652A014) and Sichuan University of Science and Engineering (2023RC10). We sincerely acknowledge the assistance of the SUSTech Core Research Facilities.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Shi, W.; Liu, C.; Lei, A. Transition-metal catalyzed oxidative cross-coupling reactions to form C–C bonds involving organometallic reagents as nucleophiles. Chem. Soc. Rev. 2011, 40, 2761-76.
2. Prier, C. K.; Rankic, D. A.; MacMillan, D. W. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013, 113, 5322-63.
3. Cherney, A. H.; Kadunce, N. T.; Reisman, S. E. Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C–C bonds. Chem. Rev. 2015, 115, 9587-652.
4. Yi, H.; Zhang, G.; Wang, H.; et al. Recent advances in radical C–H activation/radical cross-coupling. Chem. Rev. 2017, 117, 9016-85.
5. Korch, K. M.; Watson, D. A. Cross-coupling of heteroatomic electrophiles. Chem. Rev. 2019, 119, 8192-228.
6. He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J. Q. Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev. 2017, 117, 8754-86.
7. Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Pd metal catalysts for cross-couplings and related reactions in the 21st century: a critical review. Chem. Rev. 2018, 118, 2249-95.
8. Xia, Y.; Qiu, D.; Wang, J. Transition-metal-catalyzed cross-couplings through carbene migratory insertion. Chem. Rev. 2017, 117, 13810-89.
9. Takise, R.; Muto, K.; Yamaguchi, J. Cross-coupling of aromatic esters and amides. Chem. Soc. Rev. 2017, 46, 5864-88.
10. Xu, B.; Wang, Q.; Fang, C.; Zhang, Z. M.; Zhang, J. Recent advances in Pd-catalyzed asymmetric cyclization reactions. Chem. Soc. Rev. 2024, 53, 883-971.
11. Zhang, Z.; Butt, N. A.; Zhou, M.; Liu, D.; Zhang, W. Asymmetric transfer and pressure hydrogenation with earth-abundant transition metal catalysts. Chin. J. Chem. 2018, 36, 443-54.
12. Colonna, P.; Bezzenine, S.; Gil, R.; Hannedouche, J. Alkene hydroamination via earth‐abundant transition metal (iron, cobalt, copper and zinc) catalysis: a mechanistic overview. Adv. Synth. Catal. 2020, 362, 1550-63.
13. Cong, X.; Zeng, X. Mechanistic diversity of low-valent chromium catalysis: cross-coupling and hydrofunctionalization. Acc. Chem. Res. 2021, 54, 2014-26.
14. Feng, Y.; Long, S.; Tang, X.; et al. Earth-abundant 3d-transition-metal catalysts for lignocellulosic biomass conversion. Chem. Soc. Rev. 2021, 50, 6042-93.
15. Süsse, L.; Stoltz, B. M. Enantioselective formation of quaternary centers by allylic alkylation with first-row transition-metal catalysts. Chem. Rev. 2021, 121, 4084-99.
16. Cruz-navarro, J. A.; Sánchez-mora, A.; Serrano-garcía, J. S.; et al. Advances in cross-coupling reactions catalyzed by aromatic pincer complexes based on earth-abundant 3d metals (Mn, Fe, Co, Ni, Cu). Catalysts 2024, 14, 69.
17. Bansal, S.; Shabade, A. B.; Punji, B. Advances in C( sp2 )–H/C( sp2 )–H oxidative coupling of (hetero)arenes using 3d transition metal catalysts. Adv. Synth. Catal. 2021, 363, 1998-2022.
18. Narayanam, J. M.; Stephenson, C. R. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102-13.
19. Xuan, J.; Xiao, W. J. Visible-light photoredox catalysis. Angew. Chem. Int. Ed. Engl. 2012, 51, 6828-38.
21. Bellotti, P.; Huang, H. M.; Faber, T.; Glorius, F. Photocatalytic late-stage C–H functionalization. Chem. Rev. 2023, 123, 4237-352.
22. Juliá, F.; Constantin, T.; Leonori, D. Applications of halogen-atom transfer (XAT) for the generation of carbon radicals in synthetic photochemistry and photocatalysis. Chem. Rev. 2022, 122, 2292-352.
23. Ji, P.; Duan, K.; Li, M.; et al. Photochemical dearomative skeletal modifications of heteroaromatics. Chem. Soc. Rev. 2024, 53, 6600-24.
24. Zubkov, M. O.; Dilman, A. D. Radical reactions enabled by polyfluoroaryl fragments: photocatalysis and beyond. Chem. Soc. Rev. 2024, 53, 4741-85.
25. Yu, X. Y.; Chen, J. R.; Xiao, W. J. Visible light-driven radical-mediated C–C bond cleavage/functionalization in organic synthesis. Chem. Rev. 2021, 121, 506-61.
26. Skubi, K. L.; Blum, T. R.; Yoon, T. P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 2016, 116, 10035-74.
27. Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; Macmillan, D. W. C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017, 1, 0052.
28. Wang, P. Z.; Chen, J. R.; Xiao, W. J. Emerging trends in copper-promoted radical-involved C–O bond formations. J. Am. Chem. Soc. 2023, 145, 17527-50.
29. Huo, H.; Shen, X.; Wang, C.; et al. Asymmetric photoredox transition-metal catalysis activated by visible light. Nature 2014, 515, 100-3.
31. Cheung, K. P. S.; Sarkar, S.; Gevorgyan, V. Visible light-induced transition metal catalysis. Chem. Rev. 2022, 122, 1543-625.
32. Song, L.; Cai, L.; Gong, L.; Van der Eycken, E. V. Photoinduced copper-catalyzed enantioselective coupling reactions. Chem. Soc. Rev. 2023, 52, 2358-76.
33. Zhang, J.; Rueping, M. Metallaphotoredox catalysis for sp3 C–H functionalizations through single-electron transfer. Nat. Catal. 2024, 7, 963-76.
34. Zhang, J.; Rueping, M. Metallaphotoredox catalysis for sp3 C–H functionalizations through hydrogen atom transfer (HAT). Chem. Soc. Rev. 2023, 52, 4099-120.
35. Saini, V.; Stokes, B. J.; Sigman, M. S. Transition-metal-catalyzed laboratory-scale carbon–carbon bond-forming reactions of ethylene. Angew. Chem. Int. Ed. Engl. 2013, 52, 11206-20.
36. McDonald, R. I.; Liu, G.; Stahl, S. S. Palladium(II)-catalyzed alkene functionalization via nucleopalladation: stereochemical pathways and enantioselective catalytic applications. Chem. Rev. 2011, 111, 2981-3019.
37. Dhungana, R. K.; Kc, S.; Basnet, P.; Giri, R. Transition metal-catalyzed dicarbofunctionalization of unactivated olefins. Chem. Rec. 2018, 18, 1314-40.
38. Yin, G.; Mu, X.; Liu, G. Palladium(II)-catalyzed oxidative difunctionalization of alkenes: bond forming at a high-valent palladium center. Acc. Chem. Res. 2016, 49, 2413-23.
39. Wickham, L. M.; Giri, R. Transition metal (Ni, Cu, Pd)-catalyzed alkene dicarbofunctionalization reactions. Acc. Chem. Res. 2021, 54, 3415-37.
40. Qi, X.; Diao, T. Nickel-catalyzed dicarbofunctionalization of alkenes. ACS. Catal. 2020, 10, 8542-56.
41. Peng, J. Recent advances in carbonylative difunctionalization of alkenes. Adv. Synth. Catal. 2020, 362, 3059-80.
43. Wang, P. Z.; Zhang, B.; Xiao, W. J.; Chen, J. R. Photocatalysis meets copper catalysis: a new opportunity for asymmetric multicomponent radical cross-coupling reactions. Acc. Chem. Res. 2024, 57, 3433-48.
44. Engl, S.; Reiser, O. Copper-photocatalyzed ATRA reactions: concepts, applications, and opportunities. Chem. Soc. Rev. 2022, 51, 5287-99.
45. Hossain, A.; Bhattacharyya, A.; Reiser, O. Copper’s rapid ascent in visible-light photoredox catalysis. Science 2019, 364, eaav9713.
46. Abderrazak, Y.; Bhattacharyya, A.; Reiser, O. Visible-light-induced homolysis of earth-abundant metal-substrate complexes: a complementary activation strategy in photoredox catalysis. Angew. Chem. Int. Ed. Engl. 2021, 60, 21100-15.
47. Juliá, F. Ligand-to-metal charge transfer (LMCT) photochemistry at 3d-metal complexes: an emerging tool for sustainable organic synthesis. ChemCatChem 2022, 14, e202200916.
48. Kochi, J. K. Photolyses of metal compounds: cupric chloride in organic media. J. Am. Chem. Soc. 1962, 84, 2121-7.
49. Lian, P.; Long, W.; Li, J.; Zheng, Y.; Wan, X. Visible-light-induced vicinal dichlorination of alkenes through LMCT excitation of CuCl2. Angew. Chem. Int. Ed. Engl. 2020, 59, 23603-8.
50. Fumagalli, G.; Rabet, P. T.; Boyd, S.; Greaney, M. F. Three-component azidation of styrene-type double bonds: light-switchable behavior of a copper photoredox catalyst. Angew. Chem. Int. Ed. Engl. 2015, 54, 11481-4.
51. Hossain, A.; Vidyasagar, A.; Eichinger, C.; et al. Visible-light-accelerated copper(II)-catalyzed regio- and chemoselective oxo-azidation of vinyl arenes. Angew. Chem. Int. Ed. Engl. 2018, 57, 8288-92.
52. He, X. X.; Chang, H. H.; Zhao, Y. X.; et al. CuCl2 -catalyzed α-chloroketonation of aromatic alkenes via visible-light-induced LMCT. Chem. Asian. J. 2023, 18, e202200954.
53. Guo, Q.; Wang, M.; Wang, Y.; Xu, Z.; Wang, R. Photoinduced, copper-catalyzed three components cyanofluoroalkylation of alkenes with fluoroalkyl iodides as fluoroalkylation reagents. Chem. Commun. 2017, 53, 12317-20.
54. He, J.; Chen, C.; Fu, G. C.; Peters, J. C. Visible-light-induced, copper-catalyzed three-component coupling of alkyl halides, olefins, and trifluoromethylthiolate to generate trifluoromethyl thioethers. ACS. Catal. 2018, 8, 11741-8.
55. Xiong, Y.; Ma, X.; Zhang, G. Copper-catalyzed intermolecular carboamination of alkenes induced by visible light. Org. Lett. 2019, 21, 1699-703.
56. Hu, Z.; Wang, Y.; Wang, K.; Wu, J.; Wu, F. Visible-light-induced copper-catalyzed intermolecular three-component difluoroalkyl thiocyanidation of alkenes. Org. Lett. 2023, 25, 4835-9.
57. Zhang, Y.; Zhang, D. Visible-light-induced copper-catalyzed alkynylation/alkylation of alkenes. J. Org. Chem. 2020, 85, 3213-23.
58. Zhang, Y.; Sun, Y.; Chen, B.; et al. Copper-catalyzed photoinduced enantioselective dual carbofunctionalization of alkenes. Org. Lett. 2020, 22, 1490-4.
59. Yu, X. Y.; Zhao, Q. Q.; Chen, J.; Chen, J. R.; Xiao, W. J. Copper-catalyzed radical cross-coupling of redox-active oxime esters, styrenes, and boronic acids. Angew. Chem. Int. Ed. Engl. 2018, 57, 15505-9.
60. Wang, P. Z.; Zhang, Z.; Jiang, M.; Chen, J. R.; Xiao, W. J. A general copper-box system for the asymmetric arylative functionalization of benzylic, propargylic or allenylic radicals. Angew. Chem. Int. Ed. Engl. 2024, 63, e202411469.
61. Chen, J.; He, B. Q.; Wang, P. Z.; et al. Photoinduced, copper-catalyzed radical cross-coupling of cycloketone oxime esters, alkenes, and terminal alkynes. Org. Lett. 2019, 21, 4359-64.
62. Lv, X. L.; Wang, C.; Wang, Q. L.; Shu, W. Rapid synthesis of γ-arylated carbonyls enabled by the merge of copper- and photocatalytic radical relay alkylarylation of alkenes. Org. Lett. 2019, 21, 56-9.
63. Lv, X.; Shu, W. Unified and practical access to ɤ-alkynylated carbonyl derivatives via streamlined assembly at room temperature. Commun. Chem. 2019, 2, 219.
64. Wang, P. Z.; Gao, Y.; Chen, J.; Huan, X. D.; Xiao, W. J.; Chen, J. R. Asymmetric three-component olefin dicarbofunctionalization enabled by photoredox and copper dual catalysis. Nat. Commun. 2021, 12, 1815.
65. Yan, D.; Xu, S.; Qian, H.; et al. Photoredox-catalyzed and copper(II) salt-assisted radical addition/hydroxylation reaction of alkenes, sulfur ylides, and water. ACS. Catal. 2022, 12, 3279-85.
67. Kharasch, M. S.; Urry, W. H.; Jensen, E. V. Addition of derivatives of chlorinated acetic acids to olefins. J. Am. Chem. Soc. 1945, 67, 1626.
68. Kharasch, M. S.; Jensen, E. V.; Urry, W. H. Addition of carbon tetrabromide and bromoform to olefins. J. Am. Chem. Soc. 1946, 68, 154-5.
69. Wang, P.; Liang, Y.; Wu, X.; Guan, W.; Xiao, W.; Chen, J. Copper-catalyzed three-component photo-ATRA-type reaction for asymmetric intermolecular C–O coupling. ACS. Catal. 2022, 12, 10925-37.
70. Cai, Y.; Chatterjee, S.; Ritter, T. Photoinduced copper-catalyzed late-stage azidoarylation of alkenes via arylthianthrenium salts. J. Am. Chem. Soc. 2023, 145, 13542-8.
71. Zhang, B.; Li, T. T.; Mao, Z. C.; et al. Enantioselective cyanofunctionalization of aromatic alkenes via radical anions. J. Am. Chem. Soc. 2024, 146, 1410-22.
72. Wang, P. Z.; Wu, X.; Cheng, Y.; Jiang, M.; Xiao, W. J.; Chen, J. R. Photoinduced copper-catalyzed asymmetric three-component coupling of 1,3-dienes: an alternative to kharasch-sosnovsky reaction. Angew. Chem. Int. Ed. Engl. 2021, 60, 22956-62.
73. Li, G.; Meng, F.; Xiao, W.; Chen, J. Photoinduced copper-catalyzed asymmetric radical three-component cross-coupling of 1,3-enynes with oxime esters and carboxylic acids. Org. Chem. Front. 2023, 10, 2773-81.
74. Li, G. Q.; Li, Z. Q.; Jiang, M.; et al. Photoinduced copper-catalyzed asymmetric three-component radical 1,2-azidooxygenation of 1,3-dienes. Angew. Chem. Int. Ed. Engl. 2024, 63, e202405560.
75. Chen, J.; Liang, Y. J.; Wang, P. Z.; et al. Photoinduced copper-catalyzed asymmetric C–O cross-coupling. J. Am. Chem. Soc. 2021, 143, 13382-92.
76. Lu, F. D.; Lu, L. Q.; He, G. F.; Bai, J. C.; Xiao, W. J. Enantioselective radical carbocyanation of 1,3-dienes via photocatalytic generation of allylcopper complexes. J. Am. Chem. Soc. 2021, 143, 4168-73.
77. Wu, Y. L.; Jiang, M.; Rao, L.; Cheng, Y.; Xiao, W. J.; Chen, J. R. Selective three-component 1,2-aminoalkoxylation of 1-aryl-1,3-dienes by dual photoredox and copper catalysis. Org. Lett. 2022, 24, 7470-5.
78. Bi, M. H.; Cheng, Y.; Xiao, W. J.; Chen, J. R. Visible-light-induced photoredox-catalyzed selective 1,4-difluoroalkylesterification of 1-aryl-1,3-dienes. Org. Lett. 2022, 24, 7589-94.
79. Liu, Y.; Yan, H.; Chen, Y.; Hao, E.; Shi, L. Photoinduced copper-catalyzed selective three-component 1,2-amino oxygenation of 1,3-dienes. Chem. Commun. 2023, 59, 10388-91.
80. Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS. Cent. Sci. 2017, 3, 692-700.
81. García-Domínguez, A.; Mondal, R.; Nevado, C. Dual photoredox/nickel-catalyzed three-component carbofunctionalization of alkenes. Angew. Chem. Int. Ed. Engl. 2019, 58, 12286-90.
82. Campbell, M. W.; Compton, J. S.; Kelly, C. B.; Molander, G. A. Three-component olefin dicarbofunctionalization enabled by nickel/photoredox dual catalysis. J. Am. Chem. Soc. 2019, 141, 20069-78.
83. Mega, R. S.; Duong, V. K.; Noble, A.; Aggarwal, V. K. Decarboxylative conjunctive cross‐coupling of vinyl boronic esters using metallaphotoredox catalysis. Angew. Chem. Int. Ed. Engl. 2020, 132, 4405-9.
84. Sun, S. Z.; Duan, Y.; Mega, R. S.; Somerville, R. J.; Martin, R. Site-selective 1,2-dicarbofunctionalization of vinyl boronates through dual catalysis. Angew. Chem. Int. Ed. Engl. 2020, 59, 4370-4.
85. Guo, L.; Tu, H. Y.; Zhu, S.; Chu, L. Selective, intermolecular alkylarylation of alkenes via photoredox/nickel dual catalysis. Org. Lett. 2019, 21, 4771-6.
86. Li, Y.; Li, W.; Gu, Z.; Chen, J.; Xia, J. Photoredox Ni-catalyzed branch-selective reductive coupling of aldehydes with 1,3-dienes. ACS. Catal. 2020, 10, 1528-34.
87. Huang, L.; Zhu, C.; Yi, L.; Yue, H.; Kancherla, R.; Rueping, M. Cascade cross-coupling of dienes: photoredox and nickel dual catalysis. Angew. Chem. Int. Ed. Engl. 2020, 59, 457-64.
88. Zhang, Z.; Hu, X. Arylsilylation of electron-deficient alkenes via cooperative photoredox and nickel catalysis. ACS. Catal. 2020, 10, 777-82.
89. Guo, L.; Yuan, M.; Zhang, Y.; et al. General method for enantioselective three-component carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J. Am. Chem. Soc. , 2020, 20390-9.
90. Zheng, S.; Chen, Z.; Hu, Y.; et al. Selective 1,2-aryl-aminoalkylation of alkenes enabled by metallaphotoredox catalysis. Angew. Chem. 2020, 132, 18066-72.
91. Xu, S.; Chen, H.; Zhou, Z.; Kong, W. Three-component alkene difunctionalization by direct and selective activation of aliphatic C–H bonds. Angew. Chem. Int. Ed. Engl. 2021, 60, 7405-11.
92. Campbell, M. W.; Yuan, M.; Polites, V. C.; Gutierrez, O.; Molander, G. A. Photochemical C–H activation enables nickel-catalyzed olefin dicarbofunctionalization. J. Am. Chem. Soc. 2021, 143, 3901-10.
93. Jiang, H.; Yu, X.; Daniliuc, C. G.; Studer, A. Three-component aminoarylation of electron-rich alkenes by merging photoredox with nickel catalysis. Angew. Chem. Int. Ed. Engl. 2021, 60, 14399-404.
94. Qian, P.; Guan, H.; Wang, Y. E.; et al. Catalytic enantioselective reductive domino alkyl arylation of acrylates via nickel/photoredox catalysis. Nat. Commun. 2021, 12, 6613.
95. Xi, X.; Chen, Y.; Yuan, W. Nickel-catalyzed three-component alkylacylation of alkenes enabled by a photoactive electron donor-acceptor complex. Org. Lett. 2022, 24, 3938-43.
96. Dey, P.; Jana, S. K.; Rai, P.; Maji, B. Dicarbofunctionalizations of an unactivated alkene via photoredox/nickel dual catalysis. Org. Lett. 2022, 24, 6261-5.
97. Li, X.; Yuan, M.; Chen, F.; et al. Three-component enantioselective alkenylation of organophosphonates via nickel metallaphotoredox catalysis. Chem 2023, 9, 154-69.
98. Liu, M. S.; Shu, W. Rapid synthesis of β-chiral sulfones by ni-organophotocatalyzed enantioselective sulfonylalkenylation of alkenes. JACS. Au. 2023, 3, 1321-7.
99. Du, X.; Cheng-Sánchez, I.; Nevado, C. Dual nickel/photoredox-catalyzed asymmetric carbosulfonylation of alkenes. J. Am. Chem. Soc. 2023, 145, 12532-40.
100. Zhao, H.; Yuan, W. Three-component reductive conjugate addition/aldol tandem reaction enabled by nickel/photoredox dual catalysis. Chem. Sci. 2023, 14, 1485-90.
101. Ye, F.; Zheng, S.; Luo, Y.; Qi, X.; Yuan, W. Ligand-controlled regioreversed 1,2-aryl-aminoalkylation of alkenes enabled by photoredox/nickel catalysis. ACS. Catal. 2024, 14, 8505-17.
102. Koo, Y.; Hong, S. Nickel/photoredox-catalyzed three-component silylacylation of acrylates via chlorine photoelimination. Chem. Sci. 2024, 15, 7707-13.
103. Cong, F.; Sun, G. Q.; Ye, S. H.; Hu, R.; Rao, W.; Koh, M. J. A bimolecular homolytic substitution-enabled platform for multicomponent cross-coupling of unactivated alkenes. J. Am. Chem. Soc. 2024, 146, 10274-80.
104. Wang, J. Z.; Mao, E.; Nguyen, J. A.; Lyon, W. L.; MacMillan, D. W. C. Triple radical sorting: aryl-alkylation of alkenes. J. Am. Chem. Soc. 2024, 146, 15693-700.
105. Hu, X.; Cheng-Sánchez, I.; Kong, W.; Molander, G. A.; Nevado, C. Nickel-catalysed enantioselective alkene dicarbofunctionalization enabled by photochemical aliphatic C–H bond activation. Nat. Catal. 2024, 7, 655-65.
106. Schwarz, J. L.; Huang, H.; Paulisch, T. O.; Glorius, F. Dialkylation of 1,3-dienes by dual photoredox and chromium catalysis. ACS. Catal. 2020, 10, 1621-7.
107. Liu, J.; Lu, L.; Luo, Y.; et al. Photoredox-enabled chromium-catalyzed alkene diacylations. ACS. Catal. 2022, 12, 1879-85.
108. Wu, J.; Xu, X.; Duan, C.; et al. Diastereoselective 1,2-difunctionalization of 1,3-enynes enabled by merging photoexcited Hantzsch ester with chromium catalysis. Org. Chem. Front. 2024, 11, 284-9.
109. Hu, Q.; Song, S.; Zeng, T.; et al. 1,3-butadiene dicarbofunctionalization enabled by the dual role of diaryl ketone in photo-HAT/chromium catalysis. Org. Lett. 2024, 26, 1550-5.
110. Yan, H.; Zhang, D.; Liu, Y.; et al. Photoredox chromium and cobalt dual catalysis for carbonyl allylation with butadiene via allyl radical intermediates. Org. Chem. Front. 2024, 11, 684-9.
111. Li, F.; Lin, S.; Chen, Y.; et al. Photocatalytic generation of π-allyltitanium complexes via radical intermediates. Angew. Chem. Int. Ed. Engl. 2021, 60, 1561-6.
112. Li, F.; Lin, S.; Li, X.; Shi, L. Photocatalytic generation of π-allyltitanium complexes from butadiene via a radical strategy. Synthesis 2021, 53, 1889-900.
113. Hao, E.; Lu, B.; Liu, Y.; et al. Difunctionalization of 1,3-butadiene via sequential radical thiol-ene reaction and allylation by dual photoredox and titanium catalysis. Org. Lett. 2023, 25, 5094-9.
114. Yan, H.; Shan, J. R.; Zhang, F.; et al. Radical crotylation of aldehydes with 1,3-butadiene by photoredox cobalt and titanium dual catalysis. Org. Lett. 2023, 25, 7694-9.
115. Bian, K. J.; Nemoto, D. J.; Kao, S. C.; et al. Modular difunctionalization of unactivated alkenes through bio-inspired radical ligand transfer catalysis. J. Am. Chem. Soc. 2022, 144, 11810-21.
116. Li, C.; Dong, X.; Wang, Z.; Zhang, B. Visible light-initiated manganese-catalyzed hydrosulfonylation of alkenes. Green. Chem. 2023, 25, 4122-8.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].