

Metal-nitrogen-carbon catalysts towards acidic ORR in PEMFC: fundamentals, durability challenges, and improvement strategies

Abstract
Metal-Nitrogen-Carbon (M-N-C) materials are the most promising Platinum-group-metal (PGM)-free catalysts in replacing the high-cost and scarce Pt catalysts in proton exchange membrane fuel cells (PEMFCs). However, while striking improvement of M-N-C catalysts has been reached in activity, the headache degradation problems hinder their real-world application. Herein, we present a comprehensive overview of the durability of the M-N-C catalyst for oxygen reaction reduction (ORR). The fundamental understanding and identification of the ORR performance of M-N-C catalysts are discussed. Meanwhile, the standard methods to evaluate and predict the ORR performance of the PGM-free catalysts are suggested. We mainly introduce the durability challenges of the M-N-C catalyst and explain the inactivation mechanism in detail. The proposed solution and useful strategies to alleviate catalyst degradation are systematically summarized to overcome the durability bottlenecks.
Keywords
INTRODUCTION
Proton exchange membrane fuel cells (PEMFCs) are promising technologies in the upcoming hydrogen era, which could efficiently convert the chemical energy stored in H2 into electricity at high efficiency. However, the sluggish oxygen reduction reaction (ORR) process, even on the state-of-the-art Pt-based catalysts at the cathode, has greatly hampered its widespread application[1,2]. Due to costly and scarce Pt catalysts, tremendous efforts have been devoted to lowering the loading of platinum group metal (PGM) by designing PtM alloy or even to completely eliminate its usage by developing new catalyst structures, i.e., developing PGM-free catalysts[3]. The current landscape of PGM-free catalysts encompasses various materials such as M-N-C (Metal-Nitrogen-Carbon) catalysts, carbon-free support catalysts[4], and non-metal catalysts[5]. Among these, M-N-C has exhibited superior ORR performance relative to other PGM-free catalysts[6]. Furthermore, M-N-C catalysts are less expensive and exhibit excellent tolerance to various impurities and organic molecules compared to PGM catalysts[7]. The initial activity of the state-of-the-art M-N-C catalyst is even close to commercial Pt/C and has been revealed as the most promising substitute to replace PGM catalysts[8,9], with a study dating back to 1964 carried out by Jasinski et al.[10]. In the following decades, phthalocyanine was used as a precursor to synthesize M-N-C catalysts using a high-temperature pyrolysis procedure. The utilization of Fe-N-C catalysts in fuel cell tests was first carried out by Lefèvre et al. in 2009[11]. The catalysts achieved volumetric activity up to 99 A cm-3 at 0.8 V, which was a milestone during the development of M-N-C catalysts. In 2011, Proietti et al., by applying Zeolitic-imidazolate-framework (ZIF-8) as the precursor for M-N-C, made a breakthrough in catalytic activity, which even approaches the 2015 US Department of Energy target[12]. Wu et al. designed M-N-C (M = Fe, Co) catalysts with outstanding performance with a half-wave potential of only 60 mV lower than commercial Pt/C catalysts in the same year[13]. Recently, the initial activity of M-N-C catalysts was exceedingly advanced, which even approaches the commercial Pt/C catalysts[14,15] [Figure 1].

Figure 1. The best metric achieved each year of PGM-free catalysts. This figure is quoted with permission from Litster et al.[15].
Fe-N-C catalysts exhibit high ORR activity in acid media but are limited by their poor stability in PEMFC[16]. Fuel cell performances with Fe-N-C cathodes typically degrade by approximately 40% to 80% in the first 100 h of the durability test[12,17]. This phenomenon drives us to reveal the degradation mechanism of PGM-free catalysts. Several mechanisms have been proposed, including demetallation of M-N-C active sites, carbon corrosion, H2O2 and active oxygen species (ROS) attacking, protonation of surface N-groups, and water flooding[18,19]. Free radicals produced by the Fenton reaction cause carbon corrosion and demetallation, which is considered as the primary reason for severe active site loss[20,21]. The mechanisms described above are not specific to Fe-N-C catalysts but are also suitable for other M-N-C catalysts. For instance, Co-N-C catalyst exhibited lower reactivity of the Fenton reaction and higher resistance to demetallation[22]. To mitigate catalyst degradation, MN4 active sites with various metal centers have been investigated. Among these, CoN4[23], CuN4[24], CrN4[25], MnN4[26], NiN4[27], etc.[28], have demonstrated enhanced stability in comparison to FeN4. To this end, PGM-free catalysts have been rapidly advanced in the past few years and are now approaching the ultimate application target.
In this review, we first recognize the ORR mechanism and retrospect the identification of active sites of the M-N-C catalyst. Then, we concisely introduce the suggested evaluating and predicting methods of ORR activity and durability on non-precious metal catalysts. After that, we address a systematic discussion of the key stability issues of M-N-C catalyst. Based on the understanding, current strategies for durability improvement of M-N-C catalysts in PEMFC are point-by-point summarized. Finally, we outline the remaining challenges and possible future directions for improving M-N-C catalyst’s practicality. Such a timely and targeted review will help researchers to grasp the key status of M-N-C catalyst in PEMFC application for guiding future actions.
FUNDAMENTAL OF PGM-FREE ORR ELECTROCATALYSIS
The mechanism of ORR
The ORR mechanism usually includes two pathways in acid media. A direct 4e- pathway O2 reduction to
O2 dissociation:

OOH dissociation:

HOOH dissociation:


Indirect 4e- pathway with the yield of H2O2:

Multiple ORR intermediates (e.g., O*, OH*, OOH*) involve in ORR, which bind to the surface of catalysts via the O atoms. The rate-limiting step of ORR is determined by the adsorption energy of the ORR intermediates. Too strong binding will hinder the desorption of ORR intermediates, while too weak binding hampers the reaction of the intermediates. Following the Sabatier principle, the catalyst can achieve excellent ORR performance only if the intermediates interact properly with the surface of the catalyst[33-35]. Therefore, adjusting the absorption energy to the optimum value is the process of improving the catalytic activity. The rate-limiting steps and kinetic pathways of ORR remain unclear and are significantly affected by the catalytic site and environment. The pH environment can influence the structure of double layer and further affect the electron transfer process[36]. Pt-based catalysts always exhibited a higher overpotential in alkaline media than in acid media [Figure 2A]. It can be inferred that the Pt surface catalyzes the reduction of 4e- ORR by chemically adsorbing O2 in acidic media via an internal spherical electron transfer (ISET) mechanism. However, ORR proceeds through both inner-sphere electron transfer (ISET) and outer-sphere electron transfer (OSET) mechanisms in alkaline media [Figure 2B]. Notably, PGM-free catalysts generally perform better in alkaline media than in acid media, even outperforming Pt/C catalysts sometimes[37]. For PGM-free catalyst, the OSET mechanism is inhibited in alkaline media and the OH adsorption step is different with Pt/C. The OH desorption process of FeN4 site involves the reduction of (H)O-Fe3+-N4 to Fe2+-N4, which facilitates the chemisorption of O2 and the ISET mechanism. In contrast, the ISET mechanism requires more negative potential due to the lower Fe2+/3+ redox potential in acid media. This above mechanism could be used to explain PGM-free catalysts perform better in alkaline media. PGM-free catalysts behave differently in different pH environments and still need further research. Although most PGM-free catalysts have better ORR performance in alkaline media, it is meaningful to develop PGM-free catalysts with high performance in acid media due to the higher maturity of PEMFC than alkaline anion exchange membrane fuel cells[38].

Figure 2. (A) (a) Ring current measured during ORR, (b) Mass transport corrected Tafel plots; (c) ORR polarization curves in O2 saturated electrolytes; 0.1 M NaOH, and 0.1 M HClO4. (B) Schematic illustration of the double-layer structure during ORR in acidic (left) and alkaline (right) conditions. Insets (a) and (b) illustrate the ISET and OSET processes. These figures are quoted with permission from Ramaswamy et al.[36].
Identification of catalytic active sites
Understanding the structure of active sites of M-N-C catalysts is a prerequisite for designing excellent catalysts. However, synthetic procedures of M-N-C catalyst are usually involved high-temperature pyrolysis of different precursors. The complex Fe-N bond formation process and uncontrolled changes in the carbon matrix during the pyrolysis make it hard to elucidate the formation of active sites. Initially, metal particles encapsulated in carbon shells were regarded as active sites due to the presence of metal nanoparticles observed in many M-N-C catalysts[39]. With the development of advanced characterization techniques, the M-Nx structure is gradually recognized as the actual active site[40]. Lefèvre et al. first detected different ions including FeN4Cy+, FeN3Cy+, FeN2Cy+, and FeN1Cy+ by Time-of-Flight Secondary Ion Mass Spectrometry[41]. Furthermore, Chung et al. used high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) to determine the ratio of Fe and N atoms by the electron energy loss spectrum as 1:4[40], which strongly supported the MNx structure of active sites. Hitherto several types of active sites can be summarized from previous works, including MN4, MN2+2, MN5, M2N5, M-N4/M’N4, MM’N6, etc[42]. Herein, we take Fe-N-C catalyst as an example to discuss the structure-stability relationship of the active site. Recently, Jaouen et al. suggested that Fe-N-C catalysts initially contained two different FeNx sites of S1 (High-spin FeN4C12 moiety) and S2 (low- or intermediate-spin FeN4C10 moiety) by in situ 57Fe Mössbauer spectroscopy [Figure 3A][43]. While S2 sites are stable during long-time Membrane electrode assemblies (MEAs) tests, S1 sites were unstable and easily degraded to ferric oxides. They proposed two possible mechanisms for the formation of ferric oxides. S1 sites of FeNx can be directly oxidized by O2 or triggered by protonation of nitrogen and carbon surface oxidation. Following further studies on the structural evolution of the active site, Xu et al. used in situ 57Fe Mössbauer spectroscopy to reveal the relationship between the oxidized intermediates, the electric field in the electric double layer, and the Fe-N bond strength[44]. They fitted the spectrum into four doublets (note as D1, D2, D3, and D4). D1 was commonly assigned to FeN4C8 (low-spin) or FeN4C12 (high-spin) moieties. D2 was most often assigned to FeN4C10 (low or intermediate-spin) moieties. D3 might be attributed to FeN4 sites coordinating with two weak ligands. D4 might originate from the FeN4 site that adsorbed oxygen species. As shown in Figure 3B, the demetallation rate of D1 and D2 sites became faster at a high potential which corresponded to the loss of active sites density in the chronoamperometric curve, and half-wave potential in the linear sweep voltammetry (LSV) curve was more significant in relative high potential. The faster loss of D1 than D2 strongly suggested the demetallation influence of the FeN4 site. The increased content of D4 and the decreased content of D1 and D2 implied that the bare FeN4 sites could spontaneously adsorb oxygen species to form a five or sixfold-coordinate active center [Figure 3C]. Theoretical calculations demonstrated that both oxidation intermediate adsorption and electric field might lead to longer and less robust Fe-N bonds in FeN4 sites during ORR [Figure 3D]. On this basis, several new directions for improving catalyst durability were found: (1) increase the ratio and density of S2 (or D2) sites; (2) reduce the quantity of S1 or change S1 (or D1) into more stable active sites (S2); and (3) build more robust M-N bonds to reduce the effect of stretched M-N bonds.

Figure 3. (A) Coordination or structural changes of the S1 and S2 under in situ conditions. This figure is quoted with permission from Li et al.[43]. (B) The 57Fe Mössbauer spectra on Fe-N-C after degradation at different potentials for 150 h and relative contents of different types of FeN4. (C) The in situ Mössbauer spectra of the 57Fe-N-C and relative contents of different types of FeN4. (Red: D1; Purple: D2; Blue: D3; Green: D4). (D) The relationship between structural evolution and binding energy or bond length of Fe-N illustrates the bond-breaking process. These figures are quoted with permission from Xu et al.[44].
Mössbauer spectroscopy is of great importance for the structural analysis of complex Fe-N-C pyrolysis catalysts. However, the complex structure of M-N-C catalyst and limited time-space resolution characterization methods impeded the accurate identification of active moieties. The 57Fe Mössbauer spectroscopy can identify the multiplex composition in Fe-N-C catalysts, but the FeNx active sites in different oxidation and spin states still lack a theoretical basis to be distinguished[45]. Detailed analysis of various doublets from different Fe structures could not be unambiguously assigned in the experiment due to no reference materials with a single kind of active site. The differentiation of active centers and the degradation of active sites under the ORR condition are still under debate and further insights are promising gained by in situ and operando Mössbauer spectroscopy.
In short, the design of M-N-C catalysts with a single type of active site will help to analyze the structure of FeN4 sites in detail. Advanced in situ characterization techniques combined with theoretical calculations are very powerful in predicting active centers and explaining the experimental results.
Methods of evaluating ORR performance
Evaluating the ORR performance of catalysts is an essential part of designing catalysts. Only by acquiring the precise consequence can we well explain the experimental conclusions. However, accurate reflection of intrinsic ORR activity and durability remains a problem. A prevailing issue is that the catalysts exhibit excellent ORR performance in the rotating disk electrode test, but the corresponding MEA performance is still poor. The evaluation methods and test conditions can significantly influence the ORR performance. Here, we concisely discuss common methods for evaluating ORR performance from the ORR activity and ORR durability sections.
ORR activity
Highly active catalysts often suffer severe activity loss after durability testing[46-48]. However, high-durability catalysts usually have poor activity issues. The high activity of the catalysts is the precondition for durability improvement. Achieving both high activity and durability in one catalyst is our ultimate goal for practical applications. Herein, we briefly introduce the different methods for evaluating the ORR activity of catalysts. Rotating disk electrode (RDE) is the most common technique to characterize the ORR performance of the catalysts[49] and the half-wave potential is a key metric for evaluating the ORR activity of PGM-free catalysts[50-54]. The experimental operating parameters would strikingly influence the consequence. Typically, M-N-C catalysts have a catalyst loading of 4 mg/cm-2 and a thickness of 100 μm in MEAs. The catalyst loading of M-N-C catalysts ranges from 0.2-0.8 mg/cm2 in RDE and the thickness (5-20 μm) is similar to the thickness of PGM cathodes in MEAs. The higher catalyst loading usually correlates with higher half-wave potential in RDE test[53]. In contrast, the higher catalysts loading will induce more transport losses in MEA test. Thus, mass activity is also an important metric to evaluate catalytic performance[55-57]. The catalysts loading can also influence the consequence of rotating ring-disk electrode studies. The H2O2 produced from the 2-electron pathway can be reduced to water internally rather than at the ring electrode if the electrodes are thick enough[58]. Therefore, it will be more accurate to use mass activity and low loading to characterize the ORR activity and 4-electron selectivity. The electrolyte solution is also a significant parameter for ORR performance testing using RDE. The LSV curve of ORR usually measures with rotating disk electrodes in 0.1 M HClO4 and 0.5 M H2SO4 solution. The performance of M-N-C catalysts often rivals or surpasses that of Pt-based catalysts in alkaline solutions. However, their performance remains insufficient in acid solutions, due to the self-adjustment of FeN4 sites to *O-FeN4C in alkaline media[59]. Overall, the universal measurement of the RDE has been identified as simple to investigate the ORR performance of electrocatalysts.
MEAs testing and RDE testing are the most common methods to evaluate electrocatalysts activity. The MEA testing was more complicated than the RDE testing and represented practical fuel cell performances. During the test, RDE can provide a diffusion layer (5-50 µm) to simulate the fuel cell model. MEA is commonly composed of proton exchange membranes (PEM), two catalyst layers (CL), and two gas diffusion layers (GDL). Gas diffusion electrode (GDE) and catalyst coat membrane (CCM) are two typical MEA fabrication processes. Gas diffusion electrode is constructed by depositing catalyst ink on the microporous layer (MPL) of gas diffusion layers. The catalyst coat membrane is formed by depositing the catalyst ink on the membrane[60]. MEA contains a much thicker catalyst layer (5-20 µm), which is hard for mass transportation. Both O2 and H2 should pass through the flow fields, gas diffusion layers, and catalyst layers, and finally reach the catalyst surface, which will reduce the concentration of soluble gas (O2, H2)[61]. However, RDE has thinner catalyst layers (0.1-2 µm) and shorter diffusion lengths compared with MEA. Therefore, a prevalent problem with ORR catalysts was excellent activity in RDE but poor performance in the MEA testing. Translating catalyst activity and stability into MEA performance remains a challenge and requires further research[52]. Several parameters were used in fuel cell testing to evaluate catalytic performance: (1) the current density at 0.90 ViR in the H2-O2 fuel cell reflected the catalytic activity; (2) the current density at 0.80 V expressed the H2-air fuel cell performance; and (3) high current-density performance in the H2-air fuel cell in terms of a voltage at 0.8 A cm−2.
Turnover frequency (TOF) and site density (SD) are also essential indexes to evaluate the intrinsic activity of M-N-C catalyst. There are two typical methods to measure SD, including the nitrite stripping technique and the CO chemisorption technique. Primbs et al. first developed the ex situ CO cryo-adsorption and in situ reductive nitrite stripping[62]. A novel relationship was established between SD and structural information of Fe-N-C catalysts. The CO cryo-chemisorption measurement was introduced as follows: six different peak areas (△A) reflecting the amounts of CO absorption were respectively calculated from the six baseline-corrected integral pulse areas after pre-treatment, followed by six CO pluses injected into the sample loop. The calculation formula of △A was as follows:
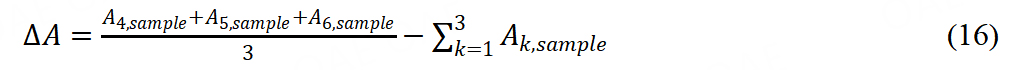
A calibration constant cf was used to convert peak areas to molar CO amounts:

The mass-based sites density with CO chemisorption could be calculated via Avogadro’s constant (NA):
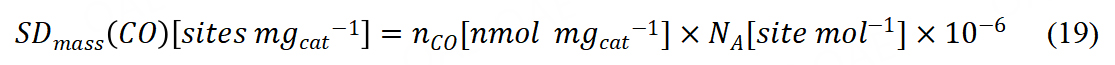
From the catalyst mass-based kinetic current, TOF could be further calculated.
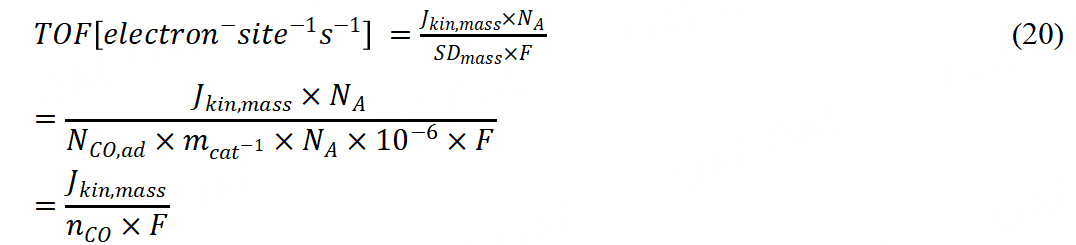
Another method was in situ reductive nitrite stripping the calculation according to:
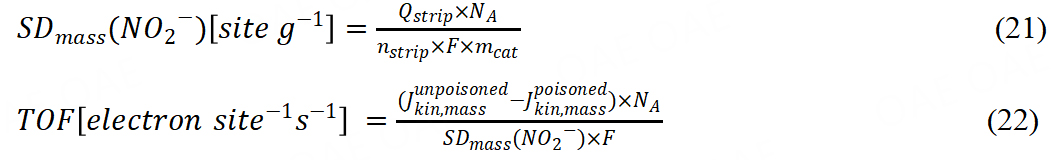
Qstrip was coulometric charge in units of coulomb associated with the NO stripping peak; nstrip was the number of electrons associated with the reduction of one nitrite ion; mcat was the mass of the catalyst; SDmass (NO2) was mass-based Fe surface site density. In recent years, Primbs et al. reported a novel in situ method for SD quantification, which employed a cyanide anion probe[62]. The mechanism was based on the relationship between cyanide-poisoned Fe3+Nx surface sites and decreased ORR activity. Irreversible poisoning of Fe3+Nx sites leads to the reduction in cyanide content, so the active sites could be quantified by ultraviolet−visible spectrophotometry. The linear correlation between cyanide concentration and absorbance supported SD and TOF calculation. It is worth noting that these different kinds of methods may not get the same SD or TOF due to the different adsorption abilities of FeNx sites. Therefore, we recommend that the same method should be used to compare the TOF values of different catalysts[63].
ORR durability
The durability test with RDE was commonly measured by the loss of half-wave potential after different cycles of the accelerated stress test (AST) (cycling the potentials from 0.6 V to 1.0 V in O2-saturated 0.5 M H2SO4). However, the MEA testing was more accurate than the RDE testing and represented practical fuel cell performances.
Currently, the PGM catalysts durability test was recommended by the US Department of Energy. AST is to cycle the cathode potential from 0.6-0.95 V in a square-wave voltage under the N2 atmosphere. The activity loss of PGM catalysts matches the loss of surface area; however, the surface area from the double-layer charging current of M-N-C cannot be used to estimate catalytic activity[64]. This is because many other factors will also cause an increase in pseudo-capacitance, such as the increased oxygenated functional groups on the carbon matrix[65]. No apparent discrepancy was observed after changing the cathode gas from N2 to O2 during the PGM catalyst durability testing. However, as for M-N-C catalysts, the potential cycle in an inert atmosphere leads to less performance loss than in oxidizing atmosphere[13]. This discrepancy might be attributed to severe carbon corrosion and demetallation caused by ROS. Therefore, the inert gas environment could not induce the same degradation process as PGM-free catalysts in the O2 atmosphere, which necessitates durability testing of PGM-free catalysts in O2 or air atmospheres. Zelenay's group reported a test protocol with standardized performance test conditions for durability testing of PGM-free ORR catalysts, which had been approved by the US Department of Energy [Table 1][66]. Herein, we summarized the RDE test [Table 2] and the relevant MEA test [Table 3] results in recent publications.
MEA test protocol of PGM-free electrocatalyst durability in air and metrics
| Test condition | Square-wave cycle in the air: steps between 0.60 V (3 s) and the OCV (but no higher than 0.925 V to minimize carbon corrosion occurring between 0.925 V and 0.95 V) (3 s); a rise time of 0.5 s or less; a single differential cell ≥ 5 cm-2; maintaining a minimum stoichiometry of ~10; a non-stabilized membrane; report fuel cell polarization curve and cathode cyclic voltammetry in N2 (or another inert gas) at time intervals and conditions specified below | |
| Total number of cycles | Until the OCV drops below 0.80 V (up to 30,000) | |
| Cycles time | 6 s < t ≤ 7 s | |
| Temperature | 80 °C | |
| Anode/cathode RH | 100%/100% | |
| Anode gas/cathode gas | H2/air | |
| Pressure | 100 kPa total partial pressure of N2 + O2 | |
| Metric | Frequency | Target |
| Catalytic activity | At the beginning and end of the test minimum | ≤ 40% loss of initial catalytic activity |
| H2-air performance at 0.80 V | After 0, 100, 1,000, 5,000, 10,000 and 30,000 cycles | 0.300 A cm-2 at the beginning of the test |
| H2-air performance at a high current density | After 0, 100, 1,000, 5,000, 10,000 and 30,000 cycles | ≤ 30 mV loss at 0.80 A cm-2 |
Summary of the RDE and MEA performance of catalysts in recent publications
| Catalysts | Active sites | RDE performance | MEA activity | ||
| E1/2 | Electrolyte | RDE test ΔE1/2 | Peak power density | ||
| P(AA-MA)(5-1)-Fe-N[67] | FeN4 | 0.843 V | 0.5 M H2SO4 | 3 mV (5k cycles) | 0.65 Wcm-2(H2- O2) |
| surf-FeN4-HPC[68] | FeN4 | 0.83 V | 0.1 M HClO4 | 20 mV (20k cycles) | 0.79 Wcm-2(H2- O2) |
| Fe-pyridinic N-C[69] | FeN4 | 0.825 V | 0.1 M HClO4 | 21 mV (20k cycles) | - |
| Co-N-C@F127[70] | CoN4 | 0.84 V | 0.5 M H2SO4 | 40 mV (30k cycles) | 0.87 Wcm-2(H2- O2) |
| Co(mIm)-NC[22] | CoN4 | 0.82 V | 0.5 M H2SO4 | 8 mV (10k cycles) | 0.64 Wcm-2(H2- O2) |
| Co-N-C-10[71] | Co2N5 | 0.79 V | 0.1 M HClO4 | 12 mV (20k cycles) | - |
| FeSA/FeAC-2DNPC[72] | FeN4 | 0.81 V | 0.5 M H2SO4 | 15 mV (10k cycles) | 0.8 Wcm-2(H2-O2) |
| Cr/N/C-950[25] | CrN4 | 0.773 V | 0.1 M HClO4 | 15 mV (20k cycles) | - |
| 17CVD/Fe-N-C-kat[73] | FeN4 | 0.835 V | 0.5 M H2SO4 | 35 mV (50k cycles) | 0.7 Wcm-2(H2- O2) |
| Fe0.5Co@HOMNCP[74] | FeCo | 0.903 V | 0.1 M KOH | ~0 mV (5k cycles) | - |
| p-FeNC@CoNC[75] | FeN4-CoN4 | 0.87 V | 0.5 M H2SO4 | 16 mV (30k cycles) | - |
| Fe/Zn-N-C[76] | FeN4-ZnN4 | 0.906 V | 0.1 M KOH | 12 mV (5k cycles) | - |
| N-Fe2MoC-GC[77] | FeNxC, MoNxC | 0.887 V | 0.1 M KOH | 16 mV (120k cycles) | 1.12 Wcm-2(H2- O2) |
| Fe-N-C + Ta-TiOx/KB[78] | FeN4 | 0.84 V | 0.5 M H2SO4 | 18 mV (10k cycles) | 0.7 Wcm-2(H2- O2) |
| NCAG/Fe-Cu[79] | FeN4, CuN4 | 0.94 V | 0.1 M KOH | 13 mV (8k cycles) | - |
| FeCoNX/C[80] | FeCoN5-OH | 0.86 V | 0.1 M HClO4 | 13 mV (5k cycles) | 0.819 Wcm-2(H2-O2) |
| Fe-AC-CVD[46] | FeN4 | 0.846 V | 0.5 M H2SO4 | 17 mV (30k cycles) | 0.535 Wcm-2(H2-air) |
| Fe-AC[46] | FeN4 | 0.915 V | 0.5 M H2SO4 | 124 mV (30k cycles) | 0.601 Wcm-2(H2-air) |
| Feg-NC/Phen[81] | FeN4 | 0.84 V | 0.1M H2SO4 | 16 mV (5k cycles) | 1.53 Wcm-2(H2-O2) |
| Cu/G[82] | CuN4 | 0.85 V | 0.1 M KOH | 10 mV (10k cycles) | - |
| Cu-SA/N-C[24] | CuN4 | 0.895 V | 0.1 M KOH | ~0 mV (5k cycles) | - |
| 20Mn-NC-second[26] | MnN4 | 0.8 V | 0.5 M H2SO4 | 17 mV (30k cycles) | 0.46 Wcm-2(H2-O2) |
| Sn-N-C[83] | SnNx | 0.73 V | 0.1 M HClO4 | - | - |
Summary MEA performance of catalysts in recent publications
| MEA durability test | ||||
| Catalysts | Test condition | Current density at | Current density at 0.8 V | Voltage at |
| target | 30k cycles AST | > 44 mA cm-2 (H2-O2) < 40% loss | > 150 mAcm-2 H2-air | < 30 mV loss |
| surf-FeN4-HPC[68] | 30k cycles AST | 24.2-n/a mA cm-2 | 87.2-20.9 mA cm-2 H2-air | 0.49-0.37 V H2-O2 |
| FeSA/FeAC-2DNPC[72] | 150 h at 0.5 V H2/air | 15-n/a mA cm-2 | 85-30 mA cm-2 H2-air | 0.42-0.30 V H2-air |
| 17CVD/Fe-N-C-kat[73] | 30k cycles AST | 27-n/a mA cm-2 | 117-64 mA cm-2 H2-air | 0.30-0.24 V H2-air |
| N-Fe2MoC-GC[77] | 30k cycles H2/O2 alkaline fuel | - | 650-400 mA cm-2 H2-O2 | 0.75-0.72 V H2-O2 |
| Fe-N-C + Ta-TiOx/KB[78] | ADTa | 17-16.5 mA cm-2 | 106-93 mA cm-2 H2-O2 | 0.62-0.57 V H2-O2 |
| Fe-AC-CVD[46] | 30k cycles AST | 33-n/a Ma cm-2 | 85-55 mA H2-air | 0.48-0.46 V H2-air |
Methods of predicting ORR performance
Current approaches to designing M-N-C catalysts mainly rely on trial and error, mostly due to the unclear relationship between the structure and performance of ORR catalysts. A theoretical understanding of the ORR catalytic process is a major requirement for rational interpretation of experimental observations and also a prerequisite for rational improving the durability of M-N-C catalysts. Here, we briefly describe the current understanding of the relationship between catalyst theory and ORR performance.
ORR activity
The d-band theory was established to combine the surface adsorption strength with the d-state of transition metals. Since all transition metals have similar broad sp-band, the difference in absorption behavior of metal surface is almost determined by the interaction between the d-band states of metals and valance states of adsorbates[84,85]. The d-band center (εd) was used to descript the average energy of the d-band and correlated with the adsorption energy and ORR activity. Cao's group presented a design principle to predict the performance of Single-atom catalysts[86]. The free energy of each step is determined by the adsorption energy of the ORR intermediate. They found the linear relationship between electronegativity and the corresponding descriptor (φ) and adsorption energy of ΔGOH* and ΔGH*. Higher φ indicates more d-sate valence electrons in the single metal atom. The φ descriptor could be used to predict the adsorption energy of intermediates and catalytic performance.
The free energy diagram of ORR was widely used to predict the rate-determining step. For a classic example, the free energy of 4e- ORR over Pt is shown in Figure 4A[87]. At U = 1.23 V (equilibrium potential), the positive free energy change (ΔG) of the OH* to H2O reaction step means that the surface of Pt would be covered by OH* and lose its catalytic activity. The highest potential for a facile reaction process was named thermodynamic limiting potential. The difference between the equilibrium potential and the limiting potential is called the theoretical overpotential (η), which is usually used to measure the activity of catalysts. The lower theoretical overpotential often corresponds to higher catalytic performance[88]. Therefore, the adsorption energy regulation of ORR intermediates was a key to rationally designing highly active catalysts. Similarly, the volcano plot was universally used to illustrate the bonding energy between the active sites and intermediates, which could be split into two legs from the apex representing the strong and weak binding

ORR durability
M-N-C catalysts have revealed huge potential in replacing PGM catalysts for ORR with respect to the initial activity, but the Achilles’ heel remained in their poor stability under an acid medium. Therefore, the universal procedures to research catalyst durability are necessary for settling the key problem. Carbon corrosion, as well as the durability of catalysts, are related to the concentration of H2O2 and ROS. The rotating ring-disk electrode test was widely used to measure the yield of H2O2. However, the generation of ROS by the Fenton reaction between Fe3+ and H2O2 leads to severe degradation of catalyst performance and is also a major factor in the degradation of PEM. The Fenton reaction is shown as follows:
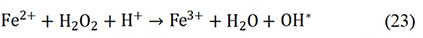
So, the content of ROS is a more significant parameter for evaluating the durability of catalysts because ROS can oxidize and destroy catalysts easily. Chen et al. used 7-hydroxyl coumarin as the probe to detect the content of OH* by analyzing the fluorescence intensity [Figure 5A][91]. Similarly, Xie et al. carried out the 6-carboxy fluorescein (6-CFL) dye as a fluorescent molecular and detected the content of ROS by comparing the fluorescence decay[78]. Moreover, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was used as a spin trap for OH* to prolong the lifetime of OH* so electron paramagnetic resonance (EPR) could detect the content of ROS. Our group proposed an in situ experiment to detect the content of ROS[25]. The mechanism is that ABTS (2, 20-azinobis (3-ethylbenzthiazoline-6-sulfonate) could react with ROS and convert to ox-ABTS which induced the color change documented by UV-vis absorption spectra [Figure 5B]. The Cr-N-C catalyst shows a much smaller change than that of the Fe-N-C system, so the stability is higher. Theoretical calculations can also be used to predict catalyst durability. Xie et al. used theoretical calculations to predict the adsorption ability of different sites for ROS and H2O2 [Figure 5C][78]. The adsorption energies of ROS and H2O2 on Fe-N-C were higher than TaO2-OH, indicating that the TaO2-OH moieties trended to be attacked by ROS and thereby protecting Fe-N-C sites.

Figure 5. (A) Schematic illustration of the conversion of non-fluorescent coumarins to fluorescent 7-hydroxycoumarins by OH*. This figure is quoted with permission from Chen et al.[91]. (B) The reaction between ROS and ABTS, and the color change of the solution after the Fenton reaction. This figure is quoted with permission from Luo et al.[25]. (C) The adsorption energy of H2O2 and ROS on the surface of Fe-N-C and TaO2-OH. This figure is quoted with permission from Xie et al.[78].
KEY STABILITY ISSUES OF M-N-C CATALYSTS
Carbon corrosion
Carbon corrosion can be primarily divided into chemical corrosion and electrochemical corrosion. These two typical types of carbon corrosion generally involve oxidation and structural damage of the carbon matrix, which are the primary degradation mechanisms of M-N-C catalysts. In general, electrochemical corrosion is always ignored because it only happens at high potential (E > 0.9 V) and the sluggish kinetic also hinders the occurrence[92]. However, in the fuel test, severe electrochemical corrosion will happen in the following situation. The absence of H2 and the presence of O2 in start-up and shut-down states result in a cathode transient potential of around 1.4 V[93]. Fuel starvation can cause a high anode potential of up to 1.6 V. The addition of oxygen evolution reaction (OER) catalysts on the electrode surface may be a feasible way to reduce the electrochemical corrosion of carbon matrix[94]. Severe carbon corrosion would lead to the collapse of the carbon matrix and falling/aggregation of active sites, which will enormously impact the activity and stability of the catalysts. Carbon corrosion could also drive the demetallation of MN4 sites next to oxidized carbon atoms, increase the mass-transport resistance around the remaining active sites, reduce the active site density and electron conductivity, and reduce the electrode porosity[28]. Therefore, carbon corrosion was regarded as the main cause of activity loss. As shown in Figure 6A, the attack of ROS eventually induced the degradation of the active sites and the formation of FeOx particles. However, another kind of mild oxidation at the carbon surface reduced the TOF of FeCxNy moieties by increasing the binding energy with O2[95]. This kind of mild oxidation was restricted to the top surface without the formation of volatile CO or CO2. In conclusion, the attack of ROS induces carbon oxidation decomposition active centers, and the leached iron ions, in turn, promote the formation of ROS from H2O2 through the Fenton reaction.

Figure 6. (A) Schematic illustration of the carbon corrosion mechanisms. This figure is quoted with permission from Wan et al.[99]. (B) Schematic demonstration of three models of Fe-N-C catalysts composed of Fe particles, Fe particles with a carbon layer, and FeNx sites. This figure is quoted with permission from Choi et al.[97].(C) Comparison of the quantity of leached metal after 200 cycles AST (O2 or Ar environment) in alkaline media. (D) The amount of dissolved Fe normalized to the applied electric charge. Relative losses of the power density (PD) at 0.65 V RHE (E) and the Fe content (F) over the Ar-AST-200, O2-AST-200, O2-AST-2000, or Ar-AST-2000 procedures. These figures are quoted with permission from Ku et al.[98].
Demetallation
The demetallation of active sites is the direct cause of activity decay during ORR. Distinguishing M-Nx/C active sites in metal/metal compounds and identifying which kind of mental leaching will directly lead to the loss of activity is the prerequisite for elucidating the demetallation mechanism during long-term ORR. Most of the Fe leaching during the ORR process originates from inactive and acid-soluble iron species such as Fe nanoparticles/oxides/sulfides/carbides[96].
Choi et al. applied a scanning flow cell connected to an inductively coupled plasma mass spectrometer (SFC/ICP-MS) to reveal the demetallation mechanism[97]. They synthesized three kinds of Fe-N-C catalysts. The first Fe-N-C catalysts included a mass of both FeNxCy active moieties and Fe particles as a reference (denoted as FeNC-wet-1). The second Fe-N-C catalysts had low amounts of Fe particles and a mass of FeNxCy active moieties (denoted as FeNC-dry-1). The third Fe-N-C catalysts contained FeNxCy active moieties and undetectable Fe particles [Figure 6B]. These different Fe particle contents in Fe-N-C catalysts were then analyzed using EXAFS and Mössbauer spectroscopy. For FeNC-wet-1 catalysts, Fe leaching could be observed evidently in the initial 20 fast cyclic voltammograms (CVs) (ca. 705 μgFegcatal−1). However, the Fe leaching amount of FeNC-dry-1and FeNC-dry-0.5 was less, ca. 87μgFegcatal−1and no more than
To elucidate the mechanism of demetallation, Xie et al. applied DFT calculation to predict the demetallation process of MN4 sites[22]. The proposed metal leaching mechanism is that the metal center of MN4 sites moves away from the state coordinating with N4 to the carbon surface and combines with two N atoms, the other two N atoms bond with H in acid media. Then the Fe center adsorbed O2 moves away from the plane and further leaches off the active sites.
Recently, Ku et al. proposed that different gas environments might induce different rates of demetallation of active sites in alkaline media[98]. By comparing the content of iron leaching in the different environments (O2, Ar), more iron leaching in the O2 environment than in the Ar environment was observed after 200 cycles AST [Figure 6C]. The linear relationship between the leaching rate of Fe and the number of electric charges indicated the correlation of current density with Fe dissolution at relatively high current densities [Figure 6D]. On the contrary, no correlation was observed between Fe dissolution and the number of potential cycles. Furthermore, the relation between the Fe leaching rate and the power density change was further discussed. Around 3.4% loss of Fe content after O2-AST-2000 induced the 34% loss of power density [Figure 6E and F]. This prominent power density in the O2 environment loss could not be fully attributed to the demetallation of FeN4 moieties, which might be related to the Fe leaching of the inactive site. An apparent decrease of the C-C signal in XPS C 1s proved that the carbon shells of Fe3C were destroyed, exposing the Fe3C moieties, which further induced the Fe leaching of Fe3C and the decrease of TOF. Fe3C might participate in the ORR process, as Jiang et al. reported[39]. The Fe3C nanoparticles might facilitate the adsorption of oxygen molecules. Thus, the coexistence of high content of FeN4 and sufficient metallic iron nanoparticles was essential for the high ORR activity. Furthermore, the S-number (the reciprocal of the charge-normalized dissolution) was used to account for charge-normalized dissolution. During the first 1500 cycles AST, the S-number stabilizes around 10-6 (around 106 electrons transfer for a leached Fe atom). The S-number increased significantly after 1500 cycles, which was consistent with the rapid decay of the initial catalytic performance and subsequent stabilization. In previous reports, the demetallation of the catalysts was widely recognized as the primary factor contributing to catalyst instability issues. The demetallation of active sites will directly induce the degradation of catalytic performance. The demetallation of inactive Fe particles will also lead to the decrease of TOF and enhanced Fenton reaction. Therefore, improving the intrinsic demetallation resistance of active sites is an effective way to improve catalyst stability.
Nitrogen protonation
The nitrogen protonation process is the N atoms of MN4 moieties combine with H to form N-H bonds, which is considered to be the rate-determining step in the degradation of the MN4 moieties[46]. After nitrogen protonation, the M-N bonds tend to break and the metal center will move away from the N4 plane and leach out during ORR. The protonation process involves attaching one or two hydrogen atoms to the surface of active moiety. Nitrogen protonation affected the C-N bond stability of the pyridinic-N doped graphene (PNG) and oxidized PNG (PNG-O). After protonation, the Fe-N bond would considerably stretch[100]. PNG and PNG-O exhibited different degrees of Fe-N bond elongation after nitrogen protonation, indicating that the adsorption of O2 could promote the stretching of the Fe-N bond. Moreover, OH* could also promote the stretching of Fe-N bonds and demetallation of active sites; if two OH* bound to the metal site on the same side, the Fe centers would form the fragile *M(OH)2 oxidation intermediates[101]. The *M(OH)2 intermediates were susceptible to successive N protonation that eventually caused irreversible damage to active sites [Figure 7]. The nitrogen protonation process further induced the demetallation and reduction of catalytic activity. Herranz et al. especially investigated N-protonation and anion-binding effects on Fe-N-C catalysts[102]. They concluded with two hypotheses. The FeN4 sites had high TOF when the active center was near the unprotonated N-groups and bound to the anions during ORR. Once the basic N-groups were protonated, the TOF of the FeN4 site would be extremely decreased. The pace of the step is dependent on the speed of polymeric chains in reaching the N-groups. In this hypothesis, the protonation of pyridinic nitrogen might be the main reason for the activity decay of PGM-free catalysts. Another hypothesis was that the FeN4 site kept high TOF when the near N-groups were protonated without anion bonds. However, the TOF of FeN4 site will decrease when the anion bond with FeN4 center. In this case, protons can rapidly access the basic N-groups, but the anions will not immediately follow with the protons. This hypothesis could be used to explain the catalytic performance of M-N-C catalysts, which exhibited high initial activity followed by a rapid decrease. In general, the nitrogen protonation process is the main reason for the decrease of TOF and the key step in the demetallation of the MN4 sites.

Figure 7. Mechanism of the demetallation induced by N protonation. This figure is quoted with permission from Yang et al.[101].
Water flooding
M-N-C catalysts with excellent performance often possess many micropores on the carbon basis in which most active sites are embedded. When the water is trapped by the micropores, the formation of three-phase interfaces is hindered and the diffusion of O2 to the active sites is difficult. This undesired process is called water flooding.
Li et al. have shown that severe water flooding occurred in thick electrode layers, which might be the main cause of the initial activity loss in the MEAs[103]. Due to the hydrophilicity of the porous carbon matrix and the chemisorption capacity of the catalyst, the micropores of the M-N-C catalyst tended to be blocked. The blocked micropores would further hinder access to oxygen and the loss of activity sites in the micropores[104]. However, Choi et al. proposed the opposite view for microporous flooding[105]. They compared the degree of micropore flooding with the observed activity loss in MEA tests. During the in situ stability test, this negligible submerged micropore was unlikely to be the main reason for performance degradation. Hence, the mechanism of micropores flooding is still unclear and needs further research.
STRATEGY FOR DURABILITY IMPROVEMENT
Stable carbon matrix
Previous studies have demonstrated that highly graphitized catalysts could significantly improve the stability of catalysts[106]. The highly graphitized carbon substrate can enhance the hydrophobicity of the catalyst, protect the catalyst from severe micropore flooding, improve the active site stability, and further improve the demetallation and corrosion resistance of the catalyst[107]. Therefore, designing a more graphitized local structure of catalysts is a feasible way to improve catalyst stability. Higher pyrolysis temperature could induce higher a degree of carbon-based graphitization[17]. Qu et al. designed Fe-Nx/C catalysts via a stepwise pyrolysis treatment which achieved a highly graphitized carbon and superior stability[108]. Only 25 mV loss of half-wave potential after 30000 cycles ADT and high retention rate of activity after 100 h fuel cell test proved the role of high levels of graphitized carbon substrates in enhancing catalyst stability. Similarly, Yan et al. designed a nitrogen-doped bimetallic carbide-graphite catalyst achieving ultra-high stability in alkaline media[77]. The catalyst (noted as N-Fe2MoC-GC) was synthesized at high temperatures (1300-1400 °C), which achieved a high degree of graphitization of carbon matrix. After the durability test, no apparently increasing in particle size in N-Fe2MoC-GC was observed. Therefore, high degree graphitization of carbon matrix could exceedingly improve the stability of catalysts. Recently, Li et al. synthesized ordered and interconnected macropores of carbon matrix [Figure 8A and Figure 8B] with FeCo alloy nanoparticles[74]. The interconnected microporous and hierarchical porous carbon matrix provided channels for mass transfer and exceedingly facilitated the exposure of active sites. The relationship between Fe and Co was elucidated by DFT calculation, and the catalysts achieved a lower energy barrier for the adsorption of intermediates. The introduction of Fe into Co induced the d-band center of Co closer to the Fermi level, which accelerated the adsorption of intermediates. Furthermore, the high stability of the catalyst was mainly due to the strong interaction of FeCo alloy nanoparticles and porous carbon matrix, and only a small amount of metal leaching was measured after the stability test. The high durability performance and minimal metal leaching of the catalysts manifested that the activity sites were firmly anchored on the ordered porous carbon matrix.

Figure 8. (A, B) Field-emission scanning electron microscopy (FESEM) image of 3D ordered porous ZIF-67 taken from different directions. These figures are quoted with permission from Li et al.[74]. Thin (C) and thick (D) carbon shell models. These figures are quoted with permission from Yoo et al.[109]. (E) Schematic demonstration of Fe-N-C catalyst with NH4Cl treatment and deposit of a carbon layer on the Fe-AC catalyst. (F, G) ORR polarization curves after different cycles AST text in O2-saturated 0.5 H2SO4. These figures are quoted with permission from Liu et al.[46] (H) H2-O2 PEMFCs polarization and power density curves. Cathode, 3.5 mgcat cm-2 for Fe-N-C; anode, 0.4 mgPt cm-2. This figure is quoted with permission from Yin et al.[81].
The carbon layer could effectively improve the durability of catalysts by preventing the catalysts from being attacked by ROS. Yoo et al. summarized the strategy of designing carbon shells on catalyst surfaces and discussed the relationship between the structure of carbon shells and the durability of catalysts[109]. On the one hand, the carbon shell provided a physical parclose that could protect the catalysts from ROS and H2O2 attack. On the other hand, the carbon shell maintained active sites and prevented metal sites from agglomeration during the ORR, thus keeping high accessible surface area. The thickness of the carbon shell was a significant influencing parameter for catalytic performance, and a thin carbon shell (< 1 nm) allowed the permeation of reactants during the ORR [Figure 8C]. However, a thick carbon shell (> 3 nm) had redundant coverage of active sites, which would hinder mass transport [Figure 8D]. Liu et al. developed a novel strategy to synthesize a more stable Fe-N-C catalyst via chemical vapor deposition (CVD), depositing an N-doped carbon layer on the Fe−N−C catalyst, resulting in excellent stability[73]. Gaseous deposition of 2-methylimidazole on Fe-doped ZnO substrates followed by thermal activation at 1000 °C to from the thin carbon layer (< 1 nm). Recently, Liu et al. reported a method for chemical vapor deposition of ZIF-8 onto Fe-AC[46]. As shown in Figure 8E, NH4Cl treatment could create amounts of defects in the carbon matrix. Further, the CVD method was used to improve the durability of the Fe-AC catalyst. On the one hand, the deposited C-N source on the catalyst could form a thin carbon layer that physically protects the active site from ROS attack. On the other hand, the CVD process could promote the transfer of S1 sites to S2 sites. As shown in Figure 8F, the Fe-AC exhibited the best ORR activity in RDE test with a half-wave potential of 0.915 V. Unfortunately, such high ORR activity decreased strikingly (E1/2: 0.915 V-0.791 V) after 30000 cycles AST test. Therefore, the CVD technique was used to solve the unstable issue. With a decrease in ORR activity, the stability of Fe-AD-CVD was increased considerably [Figure 8G]. Achieving both high activity and high durability in one catalyst remains difficult, and it is important to unravel the degradation mechanisms. The CVD technique can be used not only to deposit C-N sources to improve catalyst stability but also to deposit metal sources. Sun's group report a CVD method that deposits FeCl2 on a surface-rich pyridinic-N carbon matrix, achieving a high density and accessibility of FeN4 sites[81]. The ORR activity of Feg-NC/Phen in PEMFC even reached the DOE (2025) target with a current density of 46 mA cm-2@0.9 V and a Pmax of 1.53 Wcm-2(H2-O2), which is almost the best reported performance of M-N-C catalysts in MEA [Figure 8H]. The performance of MEAs is not only related to the ORR capability of the catalyst but also related to the MEA preparation process, catalyst ink, and test conditions. Seizing gaseous Fe2+ on the carbon matrix increased the density of surface FeN4 sites with high utilization. The introduction of defects in the carbon matrix can also improve the accessibility of the active substance. Kim et al. embedded FeCo alloy nanoparticles into a defect-rich N-doped carbon matrix[110]. The carbon matrix was oxidized by the appropriate amount of H2O2. A great number of defect sites and a high surface area carbon matrix in the catalyst improved the accessibility of active sites. The encapsulated alloy nanoparticles were protected by the carbon layer that prevented corrosion and aggregation, achieving high stability during the ORR. Therefore, regulating the structure of carbon support is essential to improve catalytic performance. The carbon matrix could not only anchor the active site on the carbon matrix but also change the electronic structure of the metal centers, which was related to the intrinsic activity of the catalysts. Therefore, designing the carbon matrix is a feasible method to improve the performance of catalysts.
Regulation of the M-Nx bonds
The length of the Fe-N bond is an essential parameter affecting the stability of catalysts. Building robust Fe-N bonds can reduce the prolongation caused by absorption of oxygenated intermediates, further increasing the resistance of demetallation. In a recent study by Ohyama et al., in situ XAS was used to analyze the durability of the 14-mcembered hexaaza macrocyclic Fe complex (Fe-14MR) [Figure 9A] and Fe phthalocyanine (FePc) [Figure 9B][111]. As shown in Figure 9C and Figure 9D, the CV peak of Fe-14MR/C was weakened to a lower degree than FePc, indicating that Fe-14MR/C was more resistant to demetallation than FePc during ORR. Furthermore, the Fe concentrations of catalysts were monitored by Fe-derived X-ray fluorescence. The Fe leaching rate of 14-membered ring ligands was much lower than FePc in acid, which also indicated the higher durability of the catalysts [Figure 9E]. Therefore, regulating the length of Fe-N bonds was key to the stability of the Fe-14MR during the ORR. It was the shorter Fe-N bond and more compact ligands that led to the high stability of Fe-14MR. Changing the binding constant and chelation of the metal center was a feasible way to regulate Fe-N bond length and further reduced the demetallation of FeN4 active sites. Increased temperature also resulted in shorter Fe-N bonds[112]. Miao et al. successfully synthesized P(AA-MA) Fe-N catalysts and regulated the lengths of Fe-N bonds by manipulating the rate of acrylic acid and maleic acid during the copolymerizing process[67]. PAA-Fe-N catalyst was used as a reference and was synthesized from homopolymerization of acrylic acid and other precursors with shorter Fe-N bonds. Fe-N bonds of different lengths for the P(AA-MA)Fe-N and PAA Fe-N catalysts were verified by XAS [Figure 9F] and Mössbauer spectroscopy. Moreover, isothermal titration calorimetry (ITC) was applied to measure the binding constant stoichiometry of ligands and metal ions. The binding constant of Fe3+ to P(AA-MA) was higher than Fe3+ with PAA or MA. As expected, the P(AA-MA) Fe-N catalysts had higher stability and activity than PAA Fe-N catalysts during the durability test. A similar consequence was achieved by Chen et al.[68]. Hierarchically porous N-doped catalysts were synthesized by pyrolysis followed by HF etching to remove Zn and SiO2 from the substrate (note as surf-FeN4-HPC). The catalysts achieved high accessibility of active sites by anchoring abundant FeN4 sites at the carbon edges. As shown in EXAFS spectra [Figure 9G], surf-FeN4-HPC had a longer length of Fe-N bonds than FeN4-HPC (anchoring on basal-plane) and achieved high durability, with only 20 mV loss of half-wave potential after 20000 cyclic voltammetry scans. These two above consequences seemed to conflict with other studies. Li et al. synthesized Fe-pyridinic N-C catalysts with a higher ratio of pyridinic N than Fe-N-C catalysts[69]. Fe-pyridinic N-C catalysts achieved both better activity and stability than Fe-N-C catalysts. DFT calculation showed that pyridine-type FeN4 had a higher Gibbs free energy during the Fe leaching process. The bond lengths of Fe-N to pyridine nitrogen and pyrrolic nitrogen were 1.88 and 2.08 Å, respectively, which also supported that the shorter Fe-N bond was stable. A similar consequence could also observe in CoN4 sites, and the Co-N-C catalysts exhibited outstanding resistance to metal leaching, which might be related to the shorter Co-N bond length[22]. The Co-N bond of Co-N-C (1.94 Å) was shorter and more stable than porphyrin-like CoN4C12 (2.01 Å)[72].

Figure 9. Schematic demonstration of the structure of Fe14MR (A) and FePc (B). CV curves for Fe-14MR/C (C) and FePc(D). (E) Relative concentration changing of Fe in Fe-14MR/C and FePc during CV cycling under N2 and O2. These figures are quoted with permission from Ohyama et al.[111]. (F) k3 -weighted FT-EXAFS spectra of P(AA-MA)(5-1)-Fe-N, PAA-Fe-N, and Fe foil samples. This figure is quoted with permission from Miao et al.[67]. (G) k3 -weighted FT-EXAFS spectra of sur-FeN4-HPC, FeN4-HPC, FePc, and Fe foil. This figure is quoted with permission from Chen et al.[68].
In a recent study by Wan et al., the introduction of Fe clusters near FeN4 sites with two-dimensional porous carbon (noted as FeSA/FeAC-2DNPC) achieved excellent catalytic performance during the ORR[72]. The strong interaction of Fe clusters and FeN4 sites could change the electronic structure of active sites and lead to a much more suitable absorption strength of ORR intermediates [Figure 10A]. The schematic of ORR is shown in Figure 10B. FeSA/FeAC-2DNPC lost only 18 mV of half-wave potential after 10,000 potential cycles in an O2-purged acidic medium. Then DFT calculation was used to elucidate the possible mechanism; the interaction between the Fe cluster and FeN4 promoted the axial adsorption of OH at the Fe center while the other side of FeN4 was used for ORR. Fe(OH)N4 achieved a lower limiting energy barrier and enhanced ORR activity. Several models were built to elucidate the catalytic performance of FeN4 sites near different Fe clusters (Fe4-N6, Fe13-N6, Fe4-C6). The different numbers of Fe atoms in the cluster have no obvious effect on the activity enhancement; on the contrary, the effect of N-coordinated Fe clusters on FeN4 centers is greater than that of C-coordinated Fe clusters. Furthermore, molecular dynamic simulation was carried out to obtain the Fe-N radial distribution function, which could be used to illustrate the Fe-N bond length [Figure 10C]. The average Fe-N bond lengths in FeN4 were 1.82-2.38 Å, which were slightly longer than that of Fe-N4/Fe4-N6 (1.82-2.22 Å), indicating that FeN4 sites near Fe4-N6 clusters are more stable than bare FeN4 sites. The Fe-N bond lengths of bare FeN4 sites were considerably stretched from 1.82-2.38 Å to 1.71-2.50 Å; on the contrary, Fe-N4/Fe4-N6 still kept compact distribution, and short Fe-N bond lengths of 1.86-2.06 Å, which correlated with the high stability of FeSA/FeAC-2DNPC, even at the elevated temperature. The compact distribution of Fe-N bonds might be attributed to the incoherent vibrations of Fe clusters and single Fe atoms. The Fe4-N6 clusters increased the stability of the active center by building robust Fe-N bonds. However, the Fe3+ from the Fe clusters seemed to contradict the mitigates of the Fenton reactions to minimize ROS generation. Notably, the FeSA/FeAC-2DNPC demonstrated higher stability relative to references, which included the catalysts containing only bare FeN4 sites (FeSA-2DNPC) and the catalysts containing Fe nanoparticles and FeN4 sites (FeSA/FeNP-2DNPC). The atomic Fe clusters were chemically bound to the carbon matrix and kept stable in acid media[113]. Some researchers reported that the metal cluster or nanoparticle in Fe-N-C catalysts improved the ORR activity in both RDE and MEA testing[114,115]. However, the unstable Fe cluster would be leaching and transferred ions, which promoted the Fenton reaction and reduced the conductivity of Nafion under the test condition[116]. Thus, the regulation M-Nx bonds by introducing metal clusters and nanoparticles should be cautious due to the degradation mechanism in PEMFCs.

Figure 10. (A) Free energy diagrams at 1.23 V for ORR over three types of active sites of Fe-N4, Fe-N4/Fe4-N6, and Fe-N4-OH/Fe4-N6. (B) Schematic ORR process on the Fe-N4 site of Fe-N4/Fe4-N6. (C) Fe-N radical distribution function of the Fe-N4 site in the models of Fe-N4 near Fe4N6 and bare Fe-N4 site at 25 °C and 80 °C. These figures are quoted with permission from Wan et al.[72].
Introduction of the fifth ligand
The demetallation of active centers may involve the adsorption of ORR intermediates in the axial direction of metal center. The introduction of the fifth ligand can reduce the elongation of M-N bond. Recent studies have shown that Fe centers could spontaneously adsorb ORR intermediates to adjust the electronic structure of the FeN4 active sites and catalyze ORR with O2 at another side[117]. Their calculation found that the Fe centers of Fe-N-C catalysts tend to be covered with OH* in wide potential rage (0.28-1.00 V) and the adsorption energy optimized by the OH* intermediates so that these sites achieved enhanced ORR performance [Figure 11A]. The theoretical result indicated that Fe(OH)N4 structure exhibited higher half-wave potential (0.88 V) and was closer to the actual result than bare FeN4 in catalyst [Figure 11B]. Our group concluded that RuN4OH structure was the actual active site in the Ru-N-C catalysts[118]. As shown in Figure 11C, RuN4OH structure achieved downhill in free energy for each step at U=0 V due to the presence of the -OH, which demonstrated facile reaction progress for ORR. The introduction of -OH downshifted the d-band center of Ru and reduced the adsorption energy for *OH intermediates. As shown in Figure 11D and Figure 11E, the linear relationship between the adsorption energy, overpotential, and Bader charge of Metals proved that the -OH ligand changed the electronic structure of the metal center, so the Ru-N-C catalysts move close to the volcano apex and achieve the high ORR performance.

Figure 11. (A) Coverages of the ORR intermediates on the Fe center as a function of U. (B) Simulated ORR polarization curves of Fe-N-C catalysts based on FeN4 and Fe(OH)N4 centers. These figures are quoted with permission from Wang et al.[117]. (C) Gibbs free energy diagram for ORR. (D) Linear relation between Bader charge of the mental and binding energy of ORR intermediates. (E) Linear relation between Bader charge of mental and overpotential. These figures are quoted with permission from Xiao et al.[118].
The MN4 site tends to be covered by molecules in the axial direction, forming a 5-coordinated or 6-coordinated structure, but these weak ligands may not keep stable in working conditions. In situ Mössbauer spectroscopy and XAS can be used to detect adsorbed species on FeN4 surfaces. Unfortunately, the time resolution is not enough to monitor the fast changes of adsorbed species on the surface of the active sites, which hinders the cognition of the relationship with axial coordination and performance. Therefore, it is necessary to develop in-situ and operando characterization techniques to resolve the dynamic mechanism and design effective electrocatalysts.
To elucidate the influence of ORR intermediates on the durability of FeN4 centers, Wei et al. used in situ spectroscopy to illustrate the interaction between FeN4 active sites and ORR intermediates[119]. During the ORR process, Fe centers would move away or toward the plane when the FeN4 site adsorbs the ORR intermediates. As shown in Figure 12A, the 593 cm−1 Raman band was attributed to the vibration of in-plane Fe-N4[120]. Notably, in acid media, the Raman band at 593 cm−1 attenuated considerably at a lower potential compared with the spectroscopy at 1.0 V, implying that the Fe centers tend to move away from the plane at a relatively lower potential. This change was reversible when the potential was kept low for a short time and quickly switched back to high potential. Conversely, this change could also become irreversible when the catalyst is kept at a low potential for a long time (> 10 min) in acid media. The discrepancy in alkaline media [Figure 12B] was also observed; the enhanced intensity at 593 cm−1 with a wide potential range matched with the higher stability of FePc/C catalysts in alkaline media than in acid media. The possible mechanism was that the axial of the FeN4 site would spontaneously adsorb a hydroxyl to form a more catalytical and stable site. The axial OH* of the Fe(OH)N4 site would desorb at low potential (< 0.5 V) in acid media, while Fe(OH)N4 remained stable over a wide range of potentials in alkaline media[40].

Figure 12. In situ Raman spectroscopy of FePc/C during ORR in (A) acid solution and (B) alkaline solution. These figures are quoted with permission from Wei et al.[119]. (C) ORR mechanism at fourfold coordination FeN4C8 and fivefold coordination FeN4C8. This figure is quoted with permission from Nematollahi et al.[121].
Recently, Nematollahi et al. reported the effect of the fifth ligand including -NH2, -OH, and -SO4 on the stability of FeN4 site[121]. In the presence of the fifth ligand, the degree of Fe-N bond elongation during ORR was significantly reduced. The fifth ligand of the Fe center might improve the resistance of demetallation. The possible mechanism was that the fifth axial ligand at the FeN4 site could pull the Fe center back into the plane rather than being pulled out of the plane by ORR intermediates [Figure 12C].
Introducing radical scavengers
The addition of radical scavengers to catalysts is an active defensive strategy to reduce the content of H2O2 and ROS. The free radicals from the Fenton reaction render catalysts and PEM less durable[122]. The chemical degradations of PEMs are mainly caused by free radical attacks. Therefore, reducing the yield of free radicals was the direct way to improve the durability of PEMFCs. Radical scavengers have been used to improve the durability of PEMs. However, few attempts have been made to introduce radical scavengers into catalysts. Cerium oxide nanoparticles have the excellent scavenging capability of radicals and are widely used radical scavengers[123,124]. Wei et al. introduced CeO2 into the Fe-N-C catalyst, which achieved reduced H2O2 production[125]. The CeO2 could catalyze the reduction of H2O2.
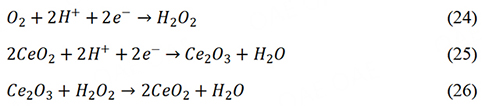
The addition of CeO2 significantly reduced the production of hydrogen peroxide and ROS, which further improved the stability of the catalyst.
Other metal oxides could also be used as radical scavengers. For example, Xie et al. reported a novel method to address the issues of carbon corrosion and demetallation, which are caused by ROS and H2O2, by introducing Ta-TiOx to Fe-N-C catalysts [Figure 13A][78], where the Ta-TiOx was used as a radical scavenger. To determine the radical scavenging ability of Ta-TiOx, 6-carboxy fluorescein (6CFL) was used as a dye to measure the content of free radicals [Figure 13B]. Fenton reagent/Ta-TiOx scavengers exhibited the lowest fluorescence decay, demonstrating the powerful scavenging ability of Ta-TiOx nanoparticles. Moreover, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was used as a spin trap for OH* to prolong the lifetime of free radicals, so electron paramagnetic resonance (EPR) could be used to further evaluate the scavenging ability. As shown in Figure 13C, the variation in EPR peak intensity of OH-DMPO with or without Ta-TiOx/KB proved that Ta-TiOx nanoparticles significantly decreased the content of free radicals. No apparent change of initial activity after the introduction of scavengers showed that the scavengers well matched up with the Fe-N-C catalysts [Figure 13D]. Ta-TiOx was introduced into Fe-N-C catalysts as a radical scavenger to catalyze the oxidation from H2O2 to O2 and H2O and scavenge radicals.

Figure 13. (A) Schematic comparing the PGM-free catalyst in the ORR process without and with the radical scavengers. (B) Stern-Volmer plot obtained using 6CFL dye in the radical solution with Fenton’s reagent, Fenton’s reagent with Fe-N-C, and Fenton’s reagent with Ta-TiOx/KB. (C) Electron paramagnetic resonance spectra of the OH-DMPO. (D) ORR performance of Fe-N-C catalysts with or without the TaTiOx-/KB radical scavengers. These figures are quoted with permission from Xie et al.[78].
Compared with the introduction of radical scavengers to catalysts, previous strategies to improve durability were the passive way to protect active sites from free radical attacks, such as increasing the degree of graphitization of carbon matrix and etc. However, the addition of radical scavenger additives was a proactive approach to eliminating H2O2 and free radicals, which further improved the stability of catalysts.
Designing a diatomic catalyst
To address the instability caused by the Fenton reaction, numerous strategies have been proposed over the past few decades, such as changing the Fe metal center to another transition metal and designing diatomic catalysts. Mn-N-C[26], Co-N-C[22,70], Cu-N-C[24,82], and Sn-N-C[83] catalysts have presented higher stability due to these metal centers with weak Fenton reactivity. Thus, designing diatomic catalysts is an effective way to improve stability and activity of catalysts. The interaction between metal and metal always involves d-orbital hybridization, electronic structure change, and charge density change[126]. In particular, the transition metals have highly variable d-orbitals and spatial geometries; thus, the introduction of another transition metal into Fe-N-C catalysts was an efficient way to regulate the electronic structure of active centers. Recently, our group reported a FeCoN5/C catalyst with Fe-Co dual-atom center sites to decrease the Fenton reaction and boost ORR activity by adjusting the electronic structure of the mental site[80]. The formation energy of FeCoN5 (-11.801 eV) was calculated, which was much lower than FeN4 (-8.48 eV) and CoN4 (-7.7 eV), implying the spontaneous formation and higher stability of diatomic centers. FeCoN5/C achieved high catalytic activity with a half-wave potential of 0.86 V and only 13 mV loss after 5000 potential cycles. Homonuclear diatomic catalysts were also a part of diatomic catalysts, our group designed Co2N5 diatomic activity sites, which exhibited higher ORR activity than CoN4 sites[71]. Regulating the neighbor atoms of Fe was an effective way to regulate the spin state of the FeN4 site. For example, He et al., by neighboring Cu atoms with Fe atoms, successfully regulated the spin state of Fe, which achieved a low H2O2 yield (1.2%) and enhanced ORR durability[79]. The interaction of different centers can be regulated via changing bond polarity, atomic distance, bonding coordination, etc. The correlated metal atoms can not only directly bind with the metal center in the first coordination shell, but also bond with the mental center via bridging atoms (O, N, S, etc.) in the second coordination shell. Indirect coordination exhibits complicated mutual effects and effective electronic structure adjustment. For example, Liu et al. built the bridge bond of Fe-O-Ti[127], and the introduction of Ti into the second coordination shell of the Fe center changed the spin state of the Fe center, resulting in enhanced ORR activity. The introduction of bridge bonds of Fe-O-Fe into catalysts also achieved enhanced catalytic activity[128]. Recently, Peng et al. reported a novel catalyst with axial coordination with N rather than another metal atom via the bridge of O atoms (noted as FeN4-O-NCR)[129]. By theoretical calculation, FeN4-O-NCR sites were thermodynamically more stable than bare FeN4 sites. From the PDOS, after the FeN4 site was axial bridging with the O atom, the 3d-band center of the Fe atom shifted to a more negative energy direction compared to bare FeN4 sites [Figure 14A]. Therefore, the appropriate absorption energy of ORR intermediates at active sites and the higher thermodynamically stability led to the enhanced ORR performance of the catalysts. CoN4 sites exhibited higher resistance to the Fenton reaction and higher stability than FeN4 sites[22]. Intergrading CoN4 and FeN4 into one catalyst may achieve improved stability while maintaining high catalytic activity. Yang et al. integrated two single-metal sites, FeNC (shell) and CoNC (core), in the catalysts (noted as p-FeNC@CoNC)[75]. From the result of Raman spectroscopy, p-FeNC@CoNC (0.93) had a medium ratio of ID/IG, implying a higher degree of graphitization carbon matrix than p-Fe-N-C (0.84). Therefore, the introduction of the CoN4 metal center could induce the formation of graphitic carbon matrix. Evidently, the p-FeNC@CoNC catalysts achieved 0.98 V of onset potential and 0.87 V of half-wave potential, only 16mV loss of half-wave potential after 30000 cycles AST in acid media [Figure 14B].

Figure 14. (A) Scaling relationships between the ηORR and d-band center of Fe-3d orbits for the FeN4-based structures (pd, pr, and gr refer to pyridinic N, pyrrolic N, and graphitic N). This figure is quoted with permission from Peng et al.[129]. (B) LSV after different cycles Potential cycling during low-potential range (0.6-1.0 V vs. RHE) p-FeNC@CoNC at 25 °C in O2-saturated 0.5 M H2SO4 solution. This figure is quoted with permission from Yang et al.[75]. (C) ΔG1*H and (D) ΔG2*H for Fe/M-N-C (M = Mn, Fe, Co, Ni, Cu, and Zn). (E) Protonation diagram of Fe/Zn-N-C. These figures are quoted with permission from Li et al.[76].
The protonation of active sites is the main reason for the decrease in activity. The active sites trend to combine with H+, leading to the decrease of TOF in acid media. Recently, Li et al. designed a durable half-metallic diatomic catalyst to alleviate the influence of protonation[76]. Six transition metals (M = Mn, Zn, Ni, Cu, Co, Fe) with more than half-filled 3d orbit were selected to separately form bimetal catalyst models with Fe. The free energies (G) of protonation (both one and two hydrogen pathways) were calculated in six bimetal models [Figure 14C and D]. Fe/Zn-N-C catalysts had the highest G of protonation, indicating that the coordination of Fe and Zn atoms achieved excellent resistance to protonation. The Fe/Zn-N-C catalysts showed remarkable durability, exhibiting a half-wave potential of 0.906 V and only 12 mV loss after 5000 potential cycles, which was lower than Fe-N-C (28 mV). Both adsorptions of O2 and H+ led to the elongation of M-N bonds, with only a slight change of Fe-N bonds compared with Zn-N bonds. However, most Zn-N bonds were broken after protonation. The FeN4 active centers were protected by the sacrifice of Zn-N bonds [Figure 14E]. Therefore, designing diatomic catalysts was the feasible way to solve the unstable issue of catalysts, including insufficient intrinsic stability, protonation of N-groups, excessive H2O2 yield, etc.
Water management
Large amounts of active sites are embedded in the micropores; thus, blocking the micropores induce high mass transfer resistance due to the prevention of O2 and H+ approach to active sites. Micropore flooding always leads to activity loss of catalysts during the MEA test. Therefore, efficient water management is significant for improving the durability of PGM-free cathode in MEA. Regulating the structure of carbon matrix is a feasible method to alleviate water flooding. The hierarchical porous structure can maximize the effect of catalysts by facilitating mass transfer and alleviating micropore flooding. The introduction of macropores and microporous can offer sufficient water-draining channels for reactants transferring and alleviating micropore flooding. To elucidate the influence microporous layer in water management, Tabe et al. developed a freezing method to monitor water distribution on the catalyst layers and further illuminate the influence of microporous layer on the performance of fuel cells and the water content of gas diffusion layers[130]. The water content was further measured by comparing the presence [Figure 15A] and absence of microporous layer [Figure 15B], where the presence of water in the catalyst layers was detected as ice. The interface of catalyst layers and gas diffusion layers provided a space for the accumulation of water. In contrast, the participation of microporous layer could boost water gasification and restrain water accumulation. The enhanced MEA performance also proved the effect of micropores [Figure 15C], which offered a buffer layer for water accumulation.

Figure 15. Schematic water distribution on a cathode with (A) or without(B) microporous layer. (C) Polarization curves for the MEAs at 35 °C operations (100% and 76% RH). These figures are quoted with permission from Tabe et al.[130]. (D) The hydrophobic group (Ar-CF3) was modified in Fe-N-C catalysts to boost the resistance to carbon corrosion and water flooding. This figure is quoted with permission from Wang et al.[133]. (E) H2-O2 PEMFCs polarization and power density curves of the cells with Pt/C@Nafion and Pt/C@SDT-Nafion measured at 80 °C and 100% RH under 150 kPa. This figure is quoted with permission from Zhang et al.[134]. (F, G) Schematic representation of ionomer distribution and gas transport at high and low RH in standard and nanofiber electrode layers. These figures are quoted with permission from Kabir et al.[135]. (H) Schematic of the improvement of ORR activity caused by the enriched protons. This figure is quoted with permission from Liu et al.[136].
The electrocatalytic reaction mainly occurs in the oxygen-water-catalyst three-phase microenvironment. The prevailing strategies for water management include increasing the density of exposed active sites and optimizing the three-phase environment by tailoring the catalyst and Nafion. Modification on carbon matrix surfaces such as -OH, -COOH (hydrophilic functional groups), or heteroatom could change the electronic structure and achieve high intrinsic activity[131]. However, the introduction of hydrophilic function groups can prolong the water retention time in micropores or even block micropores, consequently resulting in high mass transfer resistance and carbon corrosion[132]. If the carbon surface is modified with hydrophobic functional groups, hydrophobic channels can be provided to improve the durability of the catalyst. For example, Wang et al. modified the Fe-N-C catalysts with a hydrophobic group (Ar-CF3) [Figure 15D][133]. Fe-N-C-F catalysts showed a high angle of 165° and kept not-wetting in the contact angles test, while for the pristine Fe-N-C catalysts, the droplet was absorbed too quickly to measure the contact angle. The stability in H2-O2 PEMFC was assessed by potentiostatic tests, which showed that the decay of Fe-N-C-F was less than that of pristine Fe-N-C, with only a 15% decay in initial performance after 100 h at 0.6 V. After 40 h of operation at 0.5 V, the content change of Fe was measured by UV-Vis spectrophotometry. The pristine Fe-N-C changed greatly (from 1.8% to 0.38%), while only a slight change of Fe content for Fe-N-C-F (from 1.2% to 1.1%) was observed. The higher hydrophobicity of carbon matrix led to less demetallation of Fe and enhanced stability of catalysts. After introducing the hydrophobic group
In conclusion, water management is an important method of catalytic performance improvement, especially in MEAs.
CONCLUSION AND OUTLOOK
The development of M-N-C catalysts seems like a long journey approaching our ultimate target of commercializing PEMFC. Many attempts were applied to remove the barriers on the way to achieving high activity and stability of catalysts. However, as the activity of M-N-C catalysts approaches that of PGM catalysts, the stability of PGM-free catalysts remains the main obstacle to widespread application. The rational design of excellent PGM-free catalysts remains difficult as the relationship between tuning the structure and improving durability is unclear. With the development of advanced characterization techniques, especially the application of several in situ techniques over the past few decades, the identification of active sites has made great strides. Nevertheless, the degradation mechanism of the active site still requires further exploration. In this review, we summarize several possible mechanisms, including carbon corrosion, demetallation, water flooding in micropores, and nitrogen protonation. Several studies proposed that carbon corrosion and demetallation may be the main problems of activity decay. A series of endeavors should focus on overcoming these challenges. Some directions are summarized below:
Advanced in situ technology. By monitoring the evolution of the surface structure, the mechanism of the catalysis and degradation processes is partially elucidated. Advanced characterization techniques combined with theoretical calculations can be used to predict and simulate the structural evolution of active sites.
Design a carbon matrix rich in mesopores and macropores. Mesopores and macropores can increase the mass transfer rate and alleviate micropore flooding. The introduction of mesopores and macropores can prevent the active sites embedded in micropores from leaching. The leached active sites in micropores may be the main cause of initial activity loss.
Intrinsic stability improvement of active sites. Building robust M-N bonds has been proven to be a valid method for improving the intrinsic stability of catalysts. Changing the binding constant of the metal center and chelation, as well as changing the ratio of pyridinic N, were successfully used to regulate the bond length of Fe-N, which exceedingly improved the intrinsic stability of catalysts.
Non-carbon-supported catalyst. Severe carbon corrosion is unavoidable and caused by electrochemical oxidation and chemical oxidation. Several non-carbon-based electrocatalysts have been advanced in the past few years, including metal alloys[137], metal oxides[138], metal hydroxides[139], nitrides[140], carbides[141], phosphides[142], metal chalcogenides[143], and microporous materials[144]. The matrix is an essential part of catalysts, providing not only a platform to anchor active sites but also a coordinated environment, which further affects the electron structure of active centers and the durability of catalysts.
In summary, M-N-C catalysts exhibit great potential to replace PGM catalysts in fuel cell technology. Although the durability of M-N-C catalysts is still insufficient for widespread application, the catalytic performance of M-N-C catalysts has dramatically advanced in the past few decades. We hold optimistic opinions and believe that with the development of technology and theoretical knowledge, the current issues of M-N-C catalysts will be solved in the future, realizing the dramatic improvement in MEA performance. In addition to hydrogen fuel cells, M-N-C catalysts have displayed promising performance in CO2 reduction reactions (CO2RR)[145,146], hydrogen evolution reaction (HER)[147,148], nitrogen reduction reactions (NRR)[149,150] and microbial fuel cell (MFC)[151-154]. M-N-C catalysts play special roles in electrochemical energy conversion and storage.
DECLARATIONS
Authors’ contributionsPrepared the manuscript: Wang S, Chu Y, Lan C
Performed manuscript correcting: Liu C, Ge J, Xing W
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work was supported by the National Natural Science Foundation of China (21633008, 21875243, U1601211), National Science and Technology Major Project (2016YFB0101202), Jilin Province Science and Technology Development Program (20200201001JC, 20190201270JC, 20180101030JC).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
2. Debe MK. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012;486:43-51.
3. Hou J, Yang M, Ke C, et al. Platinum-group-metal catalysts for proton exchange membrane fuel cells: from catalyst design to electrode structure optimization. EnergyChem 2020;2:100023.
4. Moloudi M, Noori A, Rahmanifar MS, et al. Layered double hydroxide templated synthesis of amorphous NiCoFeB as a multifunctional electrocatalyst for overall water splitting and rechargeable Zinc–Air batteries. Adv Funct Mater 2023;13:2203002.
5. Cui P, Zhao L, Long Y, Dai L, Hu C. Carbon-based electrocatalysts for acidic oxygen reduction reaction. Angew Chem Int Ed Engl 2023:e202218269.
6. Xu MJ, Ge JJ, Liu CP, Xing W. Research progress of metal-nitrogen-carbon catalysts toward oxygen reduction reaction in changchun institute of applied chemistry. J Electrochem 2020;26:464-73.
7. Li Y, Pillai HS, Wang T, et al. High-performance ammonia oxidation catalysts for anion-exchange membrane direct ammonia fuel cells. Energy Environ Sci 2021;14:1449-60.
8. Li X, He Y, Cheng S, et al. Atomic structure evolution of pt-co binary catalysts: single metal sites versus intermetallic nanocrystals. Adv Mater 2021;33:e2106371.
9. Wan X, Liu X, Li Y, et al. Fe–N–C electrocatalyst with dense active sites and efficient mass transport for high-performance proton exchange membrane fuel cells. Nat Catal 2019;2:259-68.
11. Lefèvre M, Proietti E, Jaouen F, Dodelet JP. Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 2009;324:71-4.
12. Proietti E, Jaouen F, Lefèvre M, et al. Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat Commun 2011;2:416.
13. Wu G, More KL, Johnston CM, Zelenay P. High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 2011;332:443-7.
14. Zhang H, Chung HT, Cullen DA, et al. High-performance fuel cell cathodes exclusively containing atomically dispersed iron active sites. Energy Environ Sci 2019;12:2548-58.
15. Department of Energy. Advanced PGM-free cathode engineering for high power density and durability; 2021. Available from: https://www.hydrogen.energy.gov/pdfs/review21/fc171_litster_2021_o.pdf [Last accessed on 08 Aug 2022].
16. Liu Q, Liu X, Zheng L, Shui J. The solid-phase synthesis of an Fe-N-C electrocatalyst for high-power proton-exchange membrane fuel cells. Angew Chem Int Ed Engl 2018;57:1204-8.
17. Du L, Prabhakaran V, Xie X, Park S, Wang Y, Shao Y. Low-PGM and PGM-free catalysts for proton exchange membrane fuel cells: stability challenges and material solutions. Adv Mater 2021;33:e1908232.
18. Shao Y, Dodelet JP, Wu G, Zelenay P. PGM-free cathode catalysts for PEM fuel cells: a mini-review on stability challenges. Adv Mater 2019;31:e1807615.
19. Miao Z, Li S, Priest C, Wang T, Wu G, Li Q. Effective approaches for designing stable M-N(x) /C oxygen-reduction catalysts for proton-exchange-membrane fuel cells. Adv Mater 2022;34:e2200595.
20. Banham D, Kishimoto T, Zhou Y, et al. Critical advancements in achieving high power and stable nonprecious metal catalyst-based MEAs for real-world proton exchange membrane fuel cell applications. Sci Adv 2018;4:eaar7180.
21. Liu J, Sun X, Song P, Zhang Y, Xing W, Xu W. High-performance oxygen reduction electrocatalysts based on cheap carbon black, nitrogen, and trace iron. Adv Mater 2013;25:6879-83.
22. Xie X, He C, Li B, et al. Performance enhancement and degradation mechanism identification of a single-atom Co–N–C catalyst for proton exchange membrane fuel cells. Nat Catal 2020;3:1044-54.
23. Cheng Q, Yang L, Zou L, et al. Single cobalt atom and N codoped carbon nanofibers as highly durable electrocatalyst for oxygen reduction reaction. ACS Catal 2017;7:6864-71.
24. Qu Y, Li Z, Chen W, et al. Direct transformation of bulk copper into copper single sites via emitting and trapping of atoms. Nat Catal 2018;1:781-6.
25. Luo E, Zhang H, Wang X, et al. Single-atom Cr-N(4) sites designed for durable oxygen reduction catalysis in acid media. Angew Chem Int Ed Engl 2019;58:12469-75.
26. Li J, Chen M, Cullen DA, et al. Atomically dispersed manganese catalysts for oxygen reduction in proton-exchange membrane fuel cells. Nat Catal 2018;1:935-45.
27. Wang L, Zhang J, Zheng L, et al. Carbon black-supported FM–N–C (FM = Fe, Co, and Ni) single-atom catalysts synthesized by the self-catalysis of oxygen-coordinated ferrous metal atoms. J Mater Chem A 2020;8:13166-72.
28. He Y, Liu S, Priest C, Shi Q, Wu G. Atomically dispersed metal-nitrogen-carbon catalysts for fuel cells: advances in catalyst design, electrode performance, and durability improvement. Chem Soc Rev 2020;49:3484-524.
29. Choi CH, Choi WS, Kasian O, et al. Unraveling the nature of sites active toward hydrogen peroxide reduction in Fe-N-C catalysts. Angew Chem Int Ed Engl 2017;56:8809-12.
30. Liu M, Wang L, Zhao K, et al. Atomically dispersed metal catalysts for the oxygen reduction reaction: synthesis, characterization, reaction mechanisms and electrochemical energy applications. Energy Environ Sci 2019;12:2890-923.
31. Singh H, Zhuang S, Ingis B, Nunna BB, Lee ES. Carbon-based catalysts for oxygen reduction reaction: A review on degradation mechanisms. Carbon 2019;151:160-74.
32. Duan Z, Wang G. A first principles study of oxygen reduction reaction on a Pt(111) surface modified by a subsurface transition metal M (M = Ni, Co, or Fe). Phys Chem Chem Phys 2011;13:20178-87.
33. Han Y, Wang Y, Xu R, et al. Electronic structure engineering to boost oxygen reduction activity by controlling the coordination of the central metal. Energy Environ Sci 2018;11:2348-52.
34. Ramaswamy N, Tylus U, Jia Q, Mukerjee S. Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: linking surface science to coordination chemistry. J Am Chem Soc 2013;135:15443-9.
35. Mun Y, Lee S, Kim K, et al. Versatile strategy for tuning ORR activity of a single Fe-N(4) site by controlling electron-withdrawing/donating properties of a carbon plane. J Am Chem Soc 2019;141:6254-62.
36. Ramaswamy N, Mukerjee S. Influence of inner- and outer-sphere electron transfer mechanisms during electrocatalysis of oxygen reduction in alkaline media. J Phys Chem C 2011;115:18015-26.
37. Wang X, Liu CP, Ge JJ, Zhu JB, Xing W. Recent advances in structural regulation on non-precious metal catalysts for oxygen reduction reaction in alkaline electrolytes. J Electrochem 2022;28:2108501.
38. Mustain WE, Chatenet M, Page M, Kim YS. Durability challenges of anion exchange membrane fuel cells. Energy Environ Sci 2020;13:2805-38.
39. Jiang WJ, Gu L, Li L, et al. Understanding the high activity of Fe-N-C electrocatalysts in oxygen reduction: Fe/Fe3C nanoparticles boost the activity of Fe-N(x). J Am Chem Soc 2016;138:3570-8.
40. Chung Hoon T, Cullen David A, Higgins D, et al. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 2017;357:479-84.
41. Lefèvre M, Dodelet JP, Bertrand P. Molecular oxygen reduction in pem fuel cells: evidence for the simultaneous presence of two active sites in Fe-based catalysts. J Phys Chem B 2002;106:8705-13.
42. Shah SSA, Najam T, Bashir MS, et al. Identification of catalytic active sites for durable proton exchange membrane fuel cell: catalytic degradation and poisoning perspectives. Small 2022;18:e2106279.
43. Li J, Sougrati MT, Zitolo A, et al. Identification of durable and non-durable FeNx sites in Fe–N–C materials for proton exchange membrane fuel cells. Nat Catal 2021;4:10-9.
44. Xu X, Zhang X, Kuang Z, et al. Investigation on the demetallation of Fe-N-C for oxygen reduction reaction: the influence of structure and structural evolution of active site. Appl Catal ;309:121290.
45. Mineva T, Matanovic I, Atanassov P, et al. Understanding active sites in pyrolyzed Fe–N–C catalysts for fuel cell cathodes by bridging density functional theory calculations and 57Fe mössbauer spectroscopy. ACS Catal 2019;9:9359-71.
46. Liu S, Li C, Zachman MJ, et al. Atomically dispersed iron sites with a nitrogen–carbon coating as highly active and durable oxygen reduction catalysts for fuel cells. Nat Energy 2022;7:652-63.
47. Department of energy. electrocat: durable Mn-based PGM-free catalysts for polymer electrolyte membrane fuel cells; 2021. Available from: https://www.hydrogen.energy.gov/pdfs/review21/fc170_xu_2021_o.pdf [Last accessed on 01 Feb Aug 2023].
48. He Y, Wu G. PGM-free oxygen-reduction catalyst development for proton-exchange membrane fuel cells: challenges, solutions, and promises. Acc Mater Res 2022;3:224-36.
49. Kocha SS, Shinozaki K, Zack JW, et al. Best practices and testing protocols for benchmarking ORR activities of fuel cell electrocatalysts using rotating disk electrode. Electrocatalysis 2017;8:366-74.
50. Kong F, Cui X, Huang Y, et al. N-doped carbon electrocatalyst: marked ORR activity in acidic media without the contribution from metal sites? Angew Chem Int Ed Engl 2022;61:e202116290.
51. Zhao YX, Wen JH, Li P, et al. A "pre-division metal clusters" strategy to mediate efficient dual-active sites ORR catalyst for ultralong rechargeable ZN-air battery. Angew Chem Int Ed Engl 2023;62:e202216950.
52. Lazaridis T, Stühmeier BM, Gasteiger HA, El-sayed HA. Capabilities and limitations of rotating disk electrodes versus membrane electrode assemblies in the investigation of electrocatalysts. Nat Catal 2022;5:363-73.
54. Martens S, Asen L, Ercolano G, et al. A comparison of rotating disc electrode, floating electrode technique and membrane electrode assembly measurements for catalyst testing. Jops 2018;392:274-84.
55. Jaouen F, Herranz J, Lefèvre M, et al. Cross-laboratory experimental study of non-noble-metal electrocatalysts for the oxygen reduction reaction. ACS Appl Mater Interfaces 2009;1:1623-39.
56. Zhu Y, Sokolowski J, Song X, He Y, Mei Y, Wu G. Engineering local coordination environments of atomically dispersed and heteroatom-coordinated single metal site electrocatalysts for clean energy-conversion. Adv Energy Mater 2020;10:1902844.
57. Shi Q, Hwang S, Yang H, et al. Supported and coordinated single metal site electrocatalysts. Mater Today 2020;37:93-111.
58. Suntivich J, Gasteiger HA, Yabuuchi N, Shao-horn Y. Electrocatalytic measurement methodology of oxide catalysts using a thin-film rotating disk electrode. J Electrochem Soc 2010;157:B1263.
59. Hu X, Chen S, Chen L, et al. What is the real origin of the activity of Fe-N-C electrocatalysts in the O(2) reduction reaction? J Am Chem Soc 2022;144:18144-52.
60. Liu J, Talarposhti MR, Asset T, et al. Understanding the role of interfaces for water management in platinum group metal-free electrodes in polymer electrolyte fuel cells. ACS Appl Energy Mater 2019;2:3542-53.
61. Ito H, Maeda T, Nakano A, Takenaka H. Properties of nafion membranes under PEM water electrolysis conditions. Int J Hydrogen Energy 2011;36:10527-40.
62. Primbs M, Sun Y, Roy A, et al. Establishing reactivity descriptors for platinum group metal (PGM)-free Fe–N–C catalysts for PEM fuel cells. Energy Environ Sci 2020;13:2480-500.
63. Mehmood A, Gong M, Jaouen F, et al. High loading of single atomic iron sites in Fe–NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat Catal 2022;5:311-23.
64. Stariha S, Macauley N, Sneed BT, et al. Recent advances in catalyst accelerated stress tests for polymer electrolyte membrane fuel cells. J Electrochem Soc 2018;165:F492-501.
65. Osmieri L, Cullen DA, Chung HT, Ahluwalia RK, Neyerlin K. Durability evaluation of a Fe-N-C catalyst in polymer electrolyte fuel cell environment via accelerated stress tests. Nano Energy 2020;78:105209.
66. Zhang H, Osmieri L, Park JH, et al. Standardized protocols for evaluating platinum group metal-free oxygen reduction reaction electrocatalysts in polymer electrolyte fuel cells. Nat Catal 2022;5:455-62.
67. Miao Z, Wang X, Zhao Z, et al. Improving the stability of non-noble-metal M-N-C catalysts for proton-exchange-membrane fuel cells through M-N bond length and coordination regulation. Adv Mater 2021;33:e2006613.
68. Chen G, An Y, Liu S, et al. Highly accessible and dense surface single metal FeN 4 active sites for promoting the oxygen reduction reaction. Energy Environ Sci 2022;15:2619-28.
69. Li L, Wen Y, Han G, et al. Tailoring the stability of Fe-N-C via pyridinic nitrogen for acid oxygen reduction reaction. Chem Eng J 2022;437:135320.
70. He Y, Hwang S, Cullen DA, et al. Highly active atomically dispersed CoN 4 fuel cell cathode catalysts derived from surfactant-assisted MOFs: carbon-shell confinement strategy. Energy Environ Sci 2019;12:250-60.
71. Xiao M, Zhang H, Chen Y, et al. Identification of binuclear Co2N5 active sites for oxygen reduction reaction with more than one magnitude higher activity than single atom CoN4 site. Nano Energy 2018;46:396-403.
72. Wan X, Liu Q, Liu J, et al. Iron atom-cluster interactions increase activity and improve durability in Fe-N-C fuel cells. Nat Commun 2022;13:2963.
73. Liu S, Wang M, Yang X, et al. Chemical vapor deposition for atomically dispersed and nitrogen coordinated single metal site catalysts. Angew Chem Int Ed Engl 2020;59:21698-705.
74. Li W, Liu B, Liu D, et al. Alloying co species into ordered and interconnected macroporous carbon polyhedra for efficient oxygen reduction reaction in rechargeable zinc-air batteries. Adv Mater 2022;34:e2109605.
75. Yang X, Wang M, Zachman MJ, et al. Binary atomically dispersed metal-site catalysts with core−shell nanostructures for O 2 and CO 2 reduction reactions. Small Science 2021;1:2100046.
76. Li H, Di S, Niu P, Wang S, Wang J, Li L. A durable half-metallic diatomic catalyst for efficient oxygen reduction. Energy Environ Sci 2022;15:1601-10.
77. Yan Z, Zhang Y, Jiang Z, Jiang D, Wei W, Hu Z. Nitrogen-doped bimetallic carbide-graphite composite as highly active and extremely stable electrocatalyst for oxygen reduction reaction in alkaline media. Adv Funct Materials 2022;32:2204031.
78. Xie H, Xie X, Hu G, et al. Ta–TiOx nanoparticles as radical scavengers to improve the durability of Fe–N–C oxygen reduction catalysts. Nat Energy 2022;7:281-9.
79. He T, Chen Y, Liu Q, et al. Theory-guided regulation of FeN(4) spin state by neighboring Cu atoms for enhanced oxygen reduction electrocatalysis in flexible metal-air batteries. Angew Chem Int Ed Engl 2022;61:e202201007.
80. Xiao M, Chen Y, Zhu J, et al. Climbing the apex of the ORR volcano plot via binuclear site construction: electronic and geometric engineering. J Am Chem Soc 2019;141:17763-70.
81. Yin S, Yang S, Li G, et al. Seizing gaseous Fe2+ to densify O2-accessible Fe-N4 sites for high-performance proton exchange membrane fuel cells. Energy Environ Sci 2022;15:3033-40.
82. Han G, Zheng Y, Zhang X, et al. High loading single-atom Cu dispersed on graphene for efficient oxygen reduction reaction. Nano Energy 2019;66:104088.
83. Luo F, Roy A, Silvioli L, et al. P-block single-metal-site tin/nitrogen-doped carbon fuel cell cathode catalyst for oxygen reduction reaction. Nat Mater 2020;19:1215-23.
85. Bligaard T, Nørskov J. Ligand effects in heterogeneous catalysis and electrochemistry. Electrochim Acta 2007;52:5512-6.
86. Xu H, Cheng D, Cao D, Zeng XC. A universal principle for a rational design of single-atom electrocatalysts. Nat Catal 2018;1:339-48.
87. Kulkarni A, Siahrostami S, Patel A, Nørskov JK. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem Rev 2018;118:2302-12.
88. Man IC, Su H, Calle-vallejo F, et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 2011;3:1159-65.
89. Zagal JH, Koper MT. Reactivity descriptors for the activity of molecular MN4 catalysts for the oxygen reduction reaction. Angew Chem Int Ed Engl 2016;55:14510-21.
90. Luo E, Chu Y, Liu J, et al. Pyrolyzed M–N x catalysts for oxygen reduction reaction: progress and prospects. Energy Environ Sci 2021;14:2158-85.
91. Chen L, Yu W, Wang T, et al. Fluorescence detection of hydroxyl radical generated from oxygen reduction on Fe/N/C catalyst. Sci China Chem 2020;63:198-202.
92. Martinaiou I, Shahraei A, Grimm F, et al. Effect of metal species on the stability of Me-N-C catalysts during accelerated stress tests mimicking the start-up and shut-down conditions. Electrochim Acta 2017;243:183-96.
93. Yu Y, Li H, Wang H, Yuan X, Wang G, Pan M. A review on performance degradation of proton exchange membrane fuel cells during startup and shutdown processes: Causes, consequences, and mitigation strategies. Jops 2012;205:10-23.
94. Kumar K, Dubau L, Mermoux M, et al. On the influence of oxygen on the degradation of Fe-N-C catalysts. Angew Chem Int Ed Engl 2020;59:3235-43.
95. Choi CH, Lim H, Chung MW, et al. The achilles' heel of iron-based catalysts during oxygen reduction in an acidic medium. Energy Environ Sci 2018;11:3176-82.
96. Chung HT, Won JH, Zelenay P. Active and stable carbon nanotube/nanoparticle composite electrocatalyst for oxygen reduction. Nat Commun 2013;4:1922.
97. Choi CH, Baldizzone C, Polymeros G, et al. Minimizing operando demetallation of Fe-N-C electrocatalysts in acidic medium. ACS Catal 2016;6:3136-46.
98. Ku YP, Ehelebe K, Hutzler A, et al. Oxygen Reduction reaction in alkaline media causes iron leaching from Fe-N-C electrocatalysts. J Am Chem Soc 2022;144:9753-63.
99. Wan X, Shui J. Exploring durable single-atom catalysts for proton exchange membrane fuel cells. ACS Energy Lett 2022;7:1696-705.
100. Li J, Chen S, Yang N, et al. Ultrahigh-loading zinc single-atom catalyst for highly efficient oxygen reduction in both acidic and alkaline media. Angew Chem Int Ed Engl 2019;58:7035-9.
101. Yang N, Peng L, Li L, et al. Theoretically probing the possible degradation mechanisms of an FeNC catalyst during the oxygen reduction reaction. Chem Sci 2021;12:12476-84.
102. Herranz J, Jaouen F, Lefèvre M, et al. Unveiling N-protonation and anion-binding effects on Fe/N/C-catalysts for O(2) reduction in PEM fuel cells. J Phys Chem C Nanomater Interfaces 2011;115:16087-97.
103. Li H, Tang Y, Wang Z, et al. A review of water flooding issues in the proton exchange membrane fuel cell. Jops 2008;178:103-17.
104. Fu X, Zamani P, Choi JY, et al. In situ polymer graphenization ingrained with nanoporosity in a nitrogenous electrocatalyst boosting the performance of polymer-electrolyte-membrane fuel cells. Adv Mater 2017;29:1604456.
105. Choi J, Yang L, Kishimoto T, et al. Is the rapid initial performance loss of Fe/N/C non precious metal catalysts due to micropore flooding? Energy Environ Sci 2017;10:296-305.
106. Xia D, Yang X, Xie L, et al. Direct growth of carbon nanotubes doped with single atomic Fe-N4 active sites and neighboring graphitic nitrogen for efficient and stable oxygen reduction electrocatalysis. Adv Funct Mater 2019;29:1906174.
107. Ma Q, Jin H, Zhu J, et al. Stabilizing Fe-N-C catalysts as model for oxygen reduction reaction. Adv Sci (Weinh) 2021;8:e2102209.
108. Qu X, Han Y, Chen Y, et al. Stepwise pyrolysis treatment as an efficient strategy to enhance the stability performance of Fe-NX/C electrocatalyst towards oxygen reduction reaction and proton exchange membrane fuel cell. Appl Catal 2021;295:120311.
109. Yoo JM, Shin H, Chung DY, Sung YE. Carbon shell on active nanocatalyst for stable electrocatalysis. Acc Chem Res 2022;55:1278-89.
110. Kim K, Min K, Go Y, et al. FeCo alloy nanoparticles embedded in N-doped carbon supported on highly defective ketjenblack as effective bifunctional electrocatalysts for rechargeable Zn–air batteries. Appl Catal 2022;315:121501.
111. Ohyama J, Moriya M, Takahama R, et al. High durability of a 14-membered hexaaza macrocyclic fe complex for an acidic oxygen reduction reaction revealed by in situ XAS analysis. JACS Au 2021;1:1798-804.
112. Li J, Zhang H, Samarakoon W, et al. Thermally driven structure and performance evolution of atomically dispersed FeN(4) sites for oxygen reduction. Angew Chem Int Ed Engl 2019;58:18971-80.
113. Ye W, Chen S, Lin Y, et al. Precisely tuning the number of fe atoms in clusters on N-doped carbon toward acidic oxygen reduction reaction. Chem 2019;5:2865-78.
114. Yin SH, Yang J, Han Y, et al. Construction of highly active metal-containing nanoparticles and FeCo-N(4) composite sites for the acidic oxygen reduction reaction. Angew Chem Int Ed Engl 2020;59:21976-9.
115. Liu H, Jiang L, Khan J, et al. Decorating single-atomic Mn sites with FeMn clusters to boost oxygen reduction reaction. Angew Chem Int Ed Engl 2023;62:e202214988.
116. Choi CH, Baldizzone C, Grote JP, Schuppert AK, Jaouen F, Mayrhofer KJ. Stability of Fe-N-C catalysts in acidic medium studied by operando spectroscopy. Angew Chem Int Ed Engl 2015;54:12753-7.
117. Wang Y, Tang YJ, Zhou K. Self-adjusting activity induced by intrinsic reaction intermediate in Fe-N-C single-atom catalysts. J Am Chem Soc 2019;141:14115-9.
118. Xiao M, Gao L, Wang Y, et al. Engineering energy level of metal center: ru single-atom site for efficient and durable oxygen reduction catalysis. J Am Chem Soc 2019;141:19800-6.
119. Wei J, Xia D, Wei Y, Zhu X, Li J, Gan L. Probing the oxygen reduction reaction intermediates and dynamic active site structures of molecular and pyrolyzed Fe–N–C electrocatalysts by in situ raman spectroscopy. ACS Catal 2022;12:7811-20.
120. Chen Z, Jiang S, Kang G, Nguyen D, Schatz GC, Van Duyne RP. Operando characterization of iron phthalocyanine deactivation during oxygen reduction reaction using electrochemical tip-enhanced raman spectroscopy. J Am Chem Soc 2019;141:15684-92.
121. Nematollahi P, Barbiellini B, Bansil A, et al. Identification of a robust and durable FeN 4 C
122. Goellner V, Armel V, Zitolo A, Fonda E, Jaouen F. Degradation by hydrogen peroxide of metal-nitrogen-carbon catalysts for oxygen reduction. J Electrochem Soc 2015;162:H403-14.
123. Wang Z, Tang H, Zhang H, et al. Synthesis of Nafion/CeO2 hybrid for chemically durable proton exchange membrane of fuel cell. J Membr Sci 2012;421-422:201-10.
124. Yoon KR, Lee KA, Jo S, et al. Mussel-Inspired polydopamine-treated reinforced composite membranes with self-supported CeO
125. Wei H, Su X, Liu J, et al. A CeO2 modified phenylenediamine-based Fe/N/C with enhanced durability/stability as non-precious metal catalyst for oxygen reduction reaction. Electrochem Commun 2018;88:19-23.
126. Zhang L, Guo X, Zhang S, Huang S. Building up the “genome” of bi-atom catalysts toward efficient HER/OER/ORR. J Mater Chem A 2022;10:11600-12.
127. Liu Y, Liu X, Lv Z, et al. Tuning the spin state of the iron center by bridge-bonded Fe-O-Ti ligands for enhanced oxygen reduction. Angew Chem Int Ed Engl 2022;61:e202117617.
128. Gong L, Zhang H, Wang Y, et al. Bridge bonded oxygen ligands between approximated FeN(4) sites confer catalysts with high ORR performance. Angew Chem Int Ed Engl 2020;59:13923-8.
129. Peng L, Yang J, Yang Y, et al. Mesopore-rich Fe-N-C catalyst with FeN(4)-O-NC single-atom sites delivers remarkable oxygen reduction reaction performance in alkaline media. Adv Mater 2022;34:e2202544.
130. Tabe Y, Aoyama Y, Kadowaki K, Suzuki K, Chikahisa T. Impact of micro-porous layer on liquid water distribution at the catalyst layer interface and cell performance in a polymer electrolyte membrane fuel cell. Jops 2015;287:422-30.
131. Wang XX, Swihart MT, Wu G. Achievements, challenges and perspectives on cathode catalysts in proton exchange membrane fuel cells for transportation. Nat Catal 2019;2:578-89.
132. Forouzandeh F, Li X, Banham DW, et al. Improving the corrosion resistance of proton exchange membrane fuel cell carbon supports by pentafluorophenyl surface functionalization. Jops 2018;378:732-41.
133. Wang Y, Zhu P, Yang H, et al. Surface fluorination to boost the stability of the Fe/N/C cathode in proton exchange membrane fuel cells. ChemElectroChem 2018;5:1914-21.
134. Zhang Q, Dong S, Shao P, et al. Covalent organic framework-based porous ionomers for high-performance fuel cells. Science 2022;378:181-6.
135. Kabir S, Medina S, Wang G, Bender G, Pylypenko S, Neyerlin K. Improving the bulk gas transport of Fe-N-C platinum group metal-free nanofiber electrodes via electrospinning for fuel cell applications. Nano Energy 2020;73:104791.
136. Liu J, Ma R, Chu Y, et al. Construction and regulation of a surface protophilic environment to enhance oxygen reduction reaction electrocatalytic activity. ACS Appl Mater Interfaces 2020;12:41269-76.
137. Xia Z, Guo S. Strain engineering of metal-based nanomaterials for energy electrocatalysis. Chem Soc Rev 2019;48:3265-78.
138. Zhao Q, Yan Z, Chen C, Chen J. Spinels: controlled preparation, oxygen reduction/evolution reaction application, and beyond. Chem Rev 2017;117:10121-211.
139. Jin H, Guo C, Liu X, et al. Emerging two-dimensional nanomaterials for electrocatalysis. Chem Rev 2018;118:6337-408.
140. Deng C, He R, Shen W, Li M, Zhang T. A single-atom catalyst of cobalt supported on a defective two-dimensional boron nitride material as a promising electrocatalyst for the oxygen reduction reaction: a DFT study. Phys Chem Chem Phys 2019;21:6900-7.
141. Zhang RQ, Lee TH, Yu BD, Stampfl C, Soon A. The role of titanium nitride supports for single-atom platinum-based catalysts in fuel cell technology. Phys Chem Chem Phys 2012;14:16552-7.
142. Wang X, Bai L, Lu J, et al. Rapid activation of platinum with black phosphorus for efficient hydrogen evolution. Angew Chem Int Ed Engl 2019;58:19060-6.
143. Zhang Y, Zhou Q, Zhu J, Yan Q, Dou SX, Sun W. Nanostructured metal chalcogenides for energy storage and electrocatalysis. Adv Funct Mater 2017;27:1702317.
144. Wang HF, Chen L, Pang H, Kaskel S, Xu Q. MOF-derived electrocatalysts for oxygen reduction, oxygen evolution and hydrogen evolution reactions. Chem Soc Rev 2020;49:1414-48.
145. Wang XX, Hwang S, Pan YT, et al. Ordered Pt3Co intermetallic nanoparticles derived from metal-organic frameworks for oxygen reduction. Nano Lett 2018;18:4163-71.
146. Pan F, Li B, Sarnello E, et al. Boosting CO2 reduction on Fe-N-C with sulfur incorporation: synergistic electronic and structural engineering. Nano Energy 2020;68:104384.
147. Wang ZL, Hao XF, Jiang Z, et al. C and N hybrid coordination derived Co-C-N complex as a highly efficient electrocatalyst for hydrogen evolution reaction. J Am Chem Soc 2015;137:15070-3.
148. Zou X, Huang X, Goswami A, et al. Cobalt-embedded nitrogen-rich carbon nanotubes efficiently catalyze hydrogen evolution reaction at all pH values. Angew Chem Int Ed Engl 2014;53:4372-6.
149. Liu X, Jiao Y, Zheng Y, Jaroniec M, Qiao SZ. Building up a picture of the electrocatalytic nitrogen reduction activity of transition metal single-atom catalysts. J Am Chem Soc 2019;141:9664-72.
150. Mukherjee S, Cullen DA, Karakalos S, et al. Metal-organic framework-derived nitrogen-doped highly disordered carbon for electrochemical ammonia synthesis using N2 and H2O in alkaline electrolytes. Nano Energy 2018;48:217-26.
151. Cao B, Zhao Z, Peng L, et al. Silver nanoparticles boost charge-extraction efficiency in Shewanella microbial fuel cells. Science 2021;373:1336-40.
152. Koo B, Jung SP. Improvement of air cathode performance in microbial fuel cells by using catalysts made by binding metal-organic framework and activated carbon through ultrasonication and solution precipitation. Chem Eng J 2021;424:130388.
153. Jung S, Regan JM. Influence of external resistance on electrogenesis, methanogenesis, and anode prokaryotic communities in microbial fuel cells. Appl Environ Microbiol 2011;77:564-71.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Author Biographies




















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].