

Nanostructured heterogeneous photocatalyst materials for green synthesis of valuable chemicals

Abstract
The photocatalytic process employing nanostructured semiconductor materials has attracted great attention in energy production, CO2 reduction, and water/air purification for decades. Recently, applying heterogeneous photocatalyst for the synthesis of valuable chemicals is gradually emerging and considered as a promising process for the conversion of cheap resources (i.e., biomass derivatives, polyols, and aromatic hydrocarbons). Compared with traditional thermal catalytic approaches, the photocatalytic process provides a mild reaction condition and flexible platform (photocatalyst) that allows precise tweaking of reaction intermediates and reaction pathways, thus resulting in fine control of the selective synthesis of specialized chemicals that are challenging for thermal catalysis. In this review, we summarize recent achievements in photocatalytic synthesis of various industrial important chemicals via photo-oxidative and photo-reductive processes. The selective oxidation of alcohols and aromatics, epoxidation of alkenes, hydrogenation of gaseous molecules and hydrocarbons, and coupling reactions by means of various photocatalysts including metal oxides, supported plasmonic metal nanostructures, conjugated organic polymers, anchored homogeneous catalysts, and dye-sensitized heterostructures are discussed from a material perspective. In addition, fundamental understandings of reaction mechanisms and rational design of nanostructured photocatalysts for enhancing efficiency, selectivity, and stability are discussed in detail.

Keywords
INTRODUCTION
Heterogeneous catalysis is paramount in modern society, and different types of catalysts have been designed and utilized in various industrial applications, e.g., ammonia synthesis[1,2], Fischer-Tropsch synthesis[3], oil desulfurization[4], and fuel cells[5]. As a branch of heterogeneous catalysis, photocatalysis has received great attention over the last decades in water splitting, CO2 reduction, and water/air purification from both fundamental and application perspectives[6-11]. To drive these reactions using solar energy, various semiconductor-based materials, including metal oxides, nitrides, and sulfides[12-20], organic polymers[21,22], perovskites[23,24], and metal-organic frameworks (MOF)[25-28], with moderate bandgaps have been developed. These materials have been further engineered by means of constructing heterojunctions (i.e., semiconductor interfaces and loading of metal cocatalysts) to improve their photocatalytic performance in terms of efficiency and stability[29-36]. The boom in photocatalyst materials also leads to the exploration of employing a photocatalytic approach for synthetic applications[37-42] that may have an instantaneous impact.
The synthetic paths via photocatalysis exhibit several intrinsic advances compared with conventional thermal-catalytic approaches. While traditional catalysts accelerate reactions by means of forming instable surface adsorbed intermediate species with lower activation energy, the photocatalytic processes involve the excitation of the catalyst and the reactants to higher energy states that overcome the activation energy barriers. Therefore, corrosion-resistant reactors for harsh reaction conditions (i.e., high temperatures and pressures and strong oxidants and reductants) can be simplified for photocatalytic synthesis (i.e., ammonia synthesis, Fischer-Tropsch synthesis, and selective oxidation and hydrogenation reactions)[43-46]. However, researchers should be aware that specialized reactors with optimized light transmittance and mass transportation are required for heterogeneous photocatalysis. In fact, the photon energy is ~1.7-3 eV in the visible light region, which is sufficient to drive most catalytic reactions. Owing to the mild reaction conditions and tunable band positions of the semiconductor photocatalysts, some challenging redox reactions that require precise control of the redox potentials can be realized by photocatalytic approaches with high conversion and selectivity (i.e., epoxidation reactions and coupling reactions)[47-49]. This results in a significant reduction in energy consumption for separation processes (i.e., distillation, filtration, and sublimation), which is desirable for economic and environmental purposes. Nevertheless, a longer lifetime of the photocatalyst materials is expected due to such mild reaction conditions, which is essential for noble metal-supported systems, delicate surface engineered materials, and anchored homogeneous catalysts.
Employing homogeneous photocatalyst materials for the synthesis of value-added chemicals has been investigated for decades and received significant achievements. Metal complex redox catalysts (i.e., [Ru(bpz)3](PF6)2 and fac-Ir(ppy)3) and conjugated soluble dye molecules (i.e., Eosin Y and fluorescein) are frequently used in homogeneous photocatalysts due to their strong light absorbance in the visible light region, excellent solubility, and long excited-state lifetimes[50-53]. By controlling the reaction conditions and the chelating ligands of the redox complexes, a variety of important reactions including carbonyl (C-C) coupling, imino-pinacol (C-N) coupling, C-O coupling, cycloaddition, and alkylation and arylation of alkanes can be realized with high selectivity and stereo-regularity[54-65]. However, homogeneous photocatalytic systems are facing challenges in the cost of expensive metal complex catalysts, the instability of the polymeric catalysts, and the separation of products.
Several in-depth reviews summarize recent progress in utilizing heterogeneous photocatalyst for the synthesis of valuable chemicals from different perspectives, targeting specific reactions (i.e., partial oxidation, C-C coupling, and biomass products conversion), materials (i.e., TiO2, polyoxometalate, and supported plasmonic metal nanoparticles), and reactor design[45,46,66-74]. Miyabe and Kohtani[68] emphasized recent developments of TiO2-based photocatalysts in heterogeneous photocatalytic reductive and oxidative organic synthesis. Bloh et al.[69] summarized recent developments in heterogeneous photoredox catalysis with a special focus on materials optimization. This review addresses advances in photocatalytic synthesis from an application-orientated perspective, presenting the development in the design of nanostructured photocatalyst materials by covering up-to-date and classical works. We categorize the photocatalytic reactions into oxidative and reductive reactions, which are further subdivided into alcohol oxidation, epoxidation, oxidation of aromatics oxidative coupling, hydrogenation reactions, and reductive coupling [Scheme 1]. The design strategies, selection rules, advantages, and limitations of various nanostructured photocatalyst materials, including metal oxides, supported plasmonic metal nanostructures, organic polymers, anchored homogeneous catalysts, and dye-sensitized heterostructures, are also summarized. For oxidation reactions, the optimization of oxidation power of the photocatalyst is presented for the controlled synthesis of target products to avoid overoxidation. Correspondingly, the manipulation of photogenerated hydrogen atoms for selective reduction reactions by controlling the surface properties of the photocatalyst is discussed. In addition, fundamental understandings of reaction mechanisms for the rational design of nanostructured photocatalysts are presented. Moreover, this review presents the recent design and development of continuous reactors for scaling-up of photocatalytic processes.

Scheme 1. Categories of heterogeneous photocatalytic synthetic reactions discussed in this review and their major challenges.
NANOSIZED PHOTOCATALYSTS FOR OXIDATION REACTIONS
Alcohol oxidation
Monohydric alcohols
Monohydric and polyhydric alcohols are widely used as hole scavengers in photocatalytic hydrogen production (i.e., methanol, ethanol, glycerol, and triethanolamine)[75-77]. Bahruji et al.[76] showed that monohydric alcohols dissociate via the decarbonylation oxidation process when Pd nanoparticle (NP)-modified TiO2 is used as the photocatalyst under deaerated conditions, resulting in the formation of H2, CO2, and corresponding hydrocarbons, as demonstrated in Equations (1) and (2):
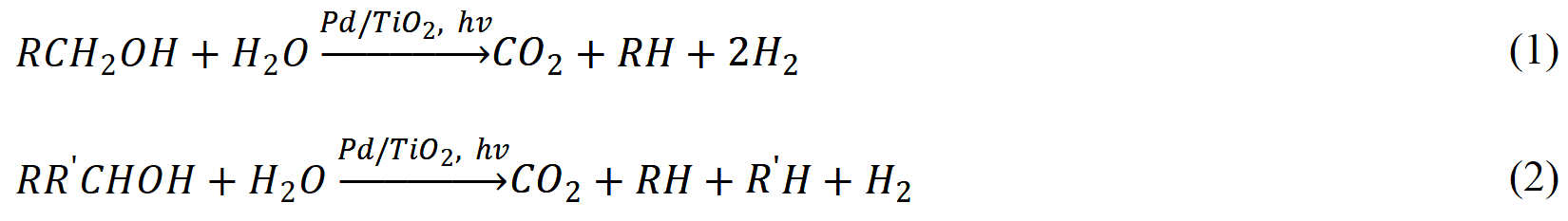
It is proposed that the alcohol decarbonylation process takes place on the Pd NP surface with CO as the reaction intermediate, which is further oxidized to CO2 by the photogenerated oxygen species on the TiO2 surface. To further enhance the activity and extend the reaction into visible light region, plasmonic metal NPs have been loaded onto various semiconductor photocatalysts (i.e., TiO2 and g-C3N4) to serve as both promoter and light absorber[78,79].
Beyond the evolution of gaseous H2, the liquid phase products may also be influenced by the identity of the metal promoter and the microstructure of the semiconductor during photocatalytic alcohol oxidation under anaerobic conditions. Ruberu et al.[80] revealed that the semiconductor-metal heterostructures dictate photocatalytic dehydrogenation and hydrogenolysis of benzyl alcohol. As demonstrated in Figure 1A, while CdS-Pt favors dehydrogenation of benzyl alcohol to generate benzaldehyde and H2, CdS0.4Se0.6-Pd favors further hydrogenolysis of the generated benzaldehyde by the photogenerated H2, resulting in the formation of toluene. A mechanistic study using labeled benzyl alcohol reveals that the adsorption of photogenerated hydrogen atoms is stronger on the Pd surface than on the Pt surface, thus resulting in the consecutive hydrogenolysis benzaldehyde. Such a design strategy is useful for synthetic chemistry via photocatalytic hydrogen transfer reactions.

Figure 1. (A) Selective oxidation of benzyl alcohol to benzaldehyde and toluene using Pt/CdS and Pd/CdS0.4Se0.6 photocatalyst, respectively. This figure is used with permission from the American Chemistry Society[80]. (B) Selective photoconversion of ethanol to butanediol using rutile TiO2 nanotubes with filled channels. This figure is used with permission from the Royal Society of Chemistry[81]. (C) Schematic demonstration of aerobic alcohol oxidation using a Pt/Ta2O5 photocatalyst under visible light irradiation. (D) Au-Pd NPs functionalized HT-PO43- for selective photocatalytic esterification of alcohols. This figure is used with permission from the American Chemistry Society[87,91]. NPs: Nanoparticles.
Lu et al.[81] showed that modulation of TiO2 microstructure and polymorph can alter the oxidation kinetics and pathway of photocatalytic ethanol reforming under deaerated conditions in water solution. While ethanol is oxidized to acetaldehyde and acetic acid over Pt-modified Degussa P25®, the value-added coupling product (2,3-butanediol) is selectively formed on rutile TiO2. The conversion of ethanol and selectivity to 2,3-butanediol can be further enhanced by employing Pt-modified rutile nanotubes with filled channels, as shown in Figure 1B. Such configuration prevents the deep oxidation of photogenerated α-hydroxyethyl radicals (CH3•CHOH) within the pores and thus facilitates the desorption of CH3•CHOH, which undergoes a coupling reaction to form 2,3-butanediol in the solution. Later, the same group reported that absolute ethanol can be directly converted to 1,1-diethoxyethane and H2 using Pt-TiO2 nanotubes and nanorods under deaerated conditions following Equations (3) and (4)[82]:
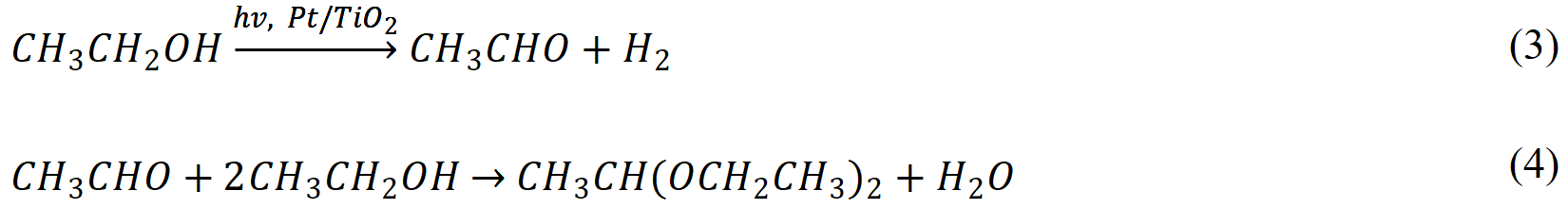
Interestingly, although almost the same photocatalyst materials were used in both studies[81,82], the liquid phase products are completely different. Therefore, we suppose that the different reaction mechanisms originate from the presence of water, which dictates the conversion of ethanol to undergo either dehydrogenation or dehydration process.
Recently, more attention has been focused on the oxidation half-reaction, where the alcohols may convert to various value-added products in liquid phase under aerobic conditions. Photocatalytic oxidation of alcohols into aldehydes or ketones has been realized under aerobic conditions using TiO2-based photocatalysts[83]; however, low efficiency and poor selectivity are the main challenges that need to be solved. Generally, loading Au, Ag, or Pt NPs on semiconductor photocatalysts can promote visible light absorption via interband transition of d band electrons and produce hot electrons (ehot-)[84], which are injected into the conduction band (CB) of the semiconductor. ehot-, together with the positive charges (holes, δ+) formed on the semiconductor, are consequently consumed by surface adsorbed O2 and alcohol molecules to complete the aerobic oxidation cycle. ehot- should not be confused with the CB electrons (eCB-) generated by semiconductor photocatalysts, as it refers to the surface localized vibrational electrons generated by plasmonic metal nanoparticles under irradiation. To further enhance the charge separation efficiency, Li et al.[85] deposited Au NPs on BiOCl with oxygen vacancies. The photogenerated ehot- can therefore be trapped at the vacancy states, thus prolonging the lifetime of the δ+ on the Au surface. However, researchers should be aware that, since the injection of ehot- into the semiconductor needs to overcome the Schottky barrier at the metal-semiconductor interface[86], the efficiency of these systems is still low in general. To further increase the efficiency, Sakamoto et al.[87] proposed depositing noble metal NPs on large bandgap semiconductors to create sufficient high work function (ϕB) and thus avoid electron transferring across the metal/semiconductor interface. Here, Pt NPs are supported on Ta2O5 (Eg ~4.0 eV) to absorb visible light. The photogenerated ehot and δ+ will be confined on the Pt surface to react with molecular O2 and alcohols, as demonstrated in Figure 1C. A series of high-performance systems have been developed with this design principle (i.e., Au/Al2O3 and Pt/SiO2) for the oxidation of various alcohols (i.e., isopropanol and benzyl alcohols) into corresponding aldehydes or ketones. Similarly, the confinement of ehot- on the metal surface can also be realized by employing engineered alloy NPs or simply metal clusters[88,89]. However, researchers should be aware that such a process is a photo-induced thermal catalytic reaction rather than a classical photocatalytic reaction.
Beyond aldehydes and ketones, alcohols can also be converted to other products via photocatalytic oxidation process. Han et al.[90] showed that Au-Ag alloy NPs supported on TiO2 can efficiently oxidize methanol into methyl formate selectively under low oxygen pressure and UV irradiation. Here, methanol gradually dissociates into methoxy (CH3O•) or formaldehyde (CH2O) on TiO2 surface. The generated formaldehyde then undergoes dehydration with another methanol molecule to generate methyl formate. The presence of metal NPs promotes oxygen dissociation to consume the surface-adsorbed H atoms into water. The optimum conversion and selectivity to methyl formate are achieved when the Au-Ag alloy with a 1:1 ratio is loaded onto TiO2. However, this reaction takes place in gas phase, and the concentration of O2 (< 0.5%) needs to be carefully controlled to avoid CO2 formation. To overcome these drawbacks, Xiao et al.[91] employed Au-Pd alloy NPs supported on phosphate-modified hydrotalcite (HT-PO43-) to convert a series of aliphatic alcohols into corresponding esters in O2 under neutral conditions. As presented in Figure 1D, phosphate anions are introduced into the calcined HT ([Mg6Al2(OH)16]CO3·mH2O) via ion exchange, which is then loaded onto Au-Pd alloy NPs. Such structure couples the basic sites of the HT support and the photocatalytic properties of the alloy NPs, which promotes the dehydrogenation of alcohols into ketones and thus the consecutive esterification of the generated ketones with alcohols via under dehydration.
Polyhydric alcohols
Catalytic conversion of polyols (i.e., diols, triols, sugars, cyclitols, and celluloses) to high-value chemicals (i.e., polyurethanes, polyesters, and polycarbonates) is of potential importance in the chemical industry and has also attracted increasing attention in photocatalysis. Employing glycerol and other polyols as hole scavengers for photocatalytic hydrogen production under deaerated conditions is the most direct application[92,93], but the conversion efficiency is relatively low. To help the design of high performance photocatalyst, Lercher’s group investigated photocatalytic reforming of a series polyols in water solution over the Rh-TiO2 system based on global kinetic analysis[94,95]. The polyol conversion takes place via three possible pathways: oxidative C-C bond cleavage from direct hole transfer to chemisorbed oxygenates on terminal Ti(IV)-OH groups, oxidation to corresponding aldoses or ketoses initiated by indirect hole trapping at surface lattice oxygen sites, and light-driven dehydration (minor). Jin et al.[96] further revealed the reaction mechanism of polyol conversion using ethylene glycol (EG) at molecular level on pristine TiO2 and metal NPs decorated TiO2 by coupling in situ surface science techniques with vibrational spectroscopies. As shown in Figure 2A and B, in situ scanning tunneling microscopy (STM) images suggest that EG molecules are adsorbed on the five-coordinated Ti (Ti5c) of rutile TiO2 (110) surface and photocatalytically converted into two parts of formaldehyde and surface-adsorbed hydrogen atoms (Hads) via oxidative C-C bond cleavage. The active sites remain on the TiO2 surface even after loading Au nanoclusters on rutile TiO2 (110), as shown in Figure 2C and D. In situ IR-MS spectroscopy suggests that the desorption of Hads to H2 is the rate-determining step (RDS), which can be promoted by metal NPs (Au, Pd, and Pt) due to the low free adsorption energy of Hads on metal surface, thus improving the dissociation of EG. Chong et al.[97] confirmed that the selectivity and efficiency in polyol conversion can be tuned by controlling the electronic and surface structure of the TiO2-based photocatalyst materials. As presented in Figure 2E-G, the selectivity in photocatalytic glycerol conversion can be tuned by varying the shape of anatase and rutile TiO2. While Pt-loaded anatase with dominant {001} or {101} facets leads to the formation of formaldehyde and CO2 with low conversion to glycolaldehyde, shaped rutile with a high percentage of {110} facets results in high selectivity towards glycolaldehyde (> 90%) without the formation of CO2.

Figure 2. (A,B) In situ STM images (2.7 × 2.7 nm) of EG adsorbed on pristine rutile (110) before and after irradiation. (C,D) In situ STM images (15 × 15 nm) of EG adsorbed on Au on rutile (110) before and after irradiation. This figure is used with permission from Elsevier[96]. (E-G) TEM images of shaped TiO2 showing tunable selectivity in glycerol conversion. (H) SEM image of the Bi2WO6 catalyst and proposed mechanism for selective oxidation of glycerol under visible-light irradiation. This figure is used with permission from the Royal Society of Chemistry[97,100]. EG: Ethylene glycol.
Photocatalytic conversion of polyols with the presence of oxygen has also been investigated. Augugliaro et al.[98] showed that most commercially available TiO2-based photocatalysts will oxidize glycerol into 1,3-dihydroxyacetone, glyceraldehyde, formic acid, and carbon dioxide in aqueous phase. Such a process requires no metal cocatalyst, as the photogenerated Hads will be consumed by the molecular oxygen to yield water. However, the selectivity in polyol conversion under oxygen conditions is more complex and difficult to control. We employed EG as the model polyol to investigate the reaction pathways and the role of metal NPs under aerated conditions using in situ IR-MS spectrometry[99]. Ethylene glycol undergoes the C-C bond cleavage process to formaldehyde, which is subsequently oxidized into CO2 or polymerized to paraformaldehyde with the photogenerated water. Similar to the reaction initiated under deaerated conditions, the removal of photogenerated Hads+ by O2 and photogenerated e- is the RDS. Interestingly, loading Ag and Au NPs on TiO2 increases the selectivity to paraformaldehyde, whereas Pt cocatalyst promotes the complete oxidation of EG to CO2. In situ IR-MS spectrometry reveals that Ag- and Au-modified TiO2 can provide water directly through Hads+ interaction with O2 and eCB-, whereas Pt/TiO2 supply water indirectly via H2 and formaldehyde oxidation.
The main drawbacks of TiO2-based photocatalysts are poor efficiency under visible light and deep oxidation of polyol into CO2. Zhang et al.[100] further developed a flower-like Bi2WO6 photocatalyst to overcome these challenges [Figure 2H]. The Bi2WO6 photocatalyst is characterized by a mild oxidation power (Eg ≈ 2.8 eV), which can selectively oxidize glycerol to 1,3-dihydroxyacetone under visible light radiation in water solution via the direct hole transfer process. Additionally, the 1,3-dihydroxyacetone molecule weakly adsorbes on Bi2WO6, which prevents deep oxidation and thus achieves high selectivity.
Although the photocatalytic oxidation of alcohols shows huge potential for the utilization of biomass as a chemical feedstock for the synthesis of value-added products, in-depth investigations are necessary to further gear up the productivity and selectivity under mild conditions. This also requires accelerating the reduction of molecular oxygen in a controlled manner for the rapid generation of ideal oxidative radical species, which is crucial in avoiding the use of extra oxidants [e.g., Hydrogen peroxide (H2O2) and O3].
Epoxidation reactions
Synthesis of epoxides via heterogeneous catalytic epoxidation of alkenes is a challenging and important reaction in both industry and fundamental research. Ag-, Au-, and Cu-based catalysts are typically employed for the selective oxidation of a series of alkenes (i.e., ethylene, propylene, and styrene), which are normally performed under elevated pressure (1-3 MPa) and moderate temperature (220 °C -280 °C) for optimized activity and selectivity[101,102]. Transition state analysis reveals that tuning the identity of metal catalyst (Ag and Cu) can promote the conversion of the common oxametallocycle intermediate to either epoxide or acetaldehyde in ethylene oxidation, thus avoiding the full oxidation of alkenes to CO2[103].
Since these metal catalysts also show plasmonic absorption in visible light region, it is reasonable to employ them as photocatalysts for epoxidation reactions under visible light irradiation. Indeed, Christopher et al.[104,105] showed that Ag nanocubes (20 wt%, ~ 75 nm) loaded on α-Al2O3 support can realize selective oxidation of ethylene to ethylene oxide with a selectivity of ~50%. Although the photocatalytic epoxidation reaction still needs to be operated at elevated temperature to achieve reasonable quantum efficiency (~40% at 200 °C and ~4% at 150 °C), the epoxidation rate is significantly higher than that of the pure thermal catalytic process. This suggests that the process is more likely a photo-induced thermal process rather than a photocatalytic approach. Later, the same group further developed Cu NPs (2 wt%, ~ 45 nm) supported on SiO2 for the selective oxidation of propylene[106]. Interestingly, such a system shows a higher selectivity in propylene epoxidation under visible light irradiation than that of the thermal process; thus, it is a photothermic-catalytic process. It is proposed that the high selectivity under irradiation is accompanied by light-induced redox of the surface Cu2O layer to metallic Cu via localized surface plasmon resonance of the Cu core.
Zhang et al.[107] employed SERS coupled with theoretical calculations to understand the active sites of individual Ag NP catalyst for plasmon excitation-assisted ethylene epoxidation. It shows that graphene layers are first generated on the Ag surface under irradiation, which in turn promote the epoxidation of ethylene at the edge defects sites. As shown in Figure 3A, TEM image reveals the activation of Ag/Al2O3 photocatalyst with in situ generated graphene layers on Ag NPs under irradiation with the presence C2H4 (A-Ag/Al2O3). Based on this evidence, they further developed a graphene/Ag/α-Al2O3 photocatalyst (G-Ag/Al2O3), which shows a similar feature in SERS spectra with distinct D and G bands of graphene compared with that of A-Ag/Al2O3 [Figure 3B]. Both the composite and the activated photocatalyst exhibit excellent performance in ethylene photo-epoxidation under ambient conditions compared with that of the conventional Ag/α-Al2O3 catalyst [Figure 3C].

Figure 3. (A,B) TEM image and SERS showing graphene formation on Ag NPs during photo-epoxidation of C2H4. (C) TOF of C2H4 epoxidation using Ag-based photocatalyst. This figure is used with permission from A Creative Commons Attribution License[107]. (D,E) TEM image of single crystal Cu NP supported on TiN and the proposed reaction mechanisms in photo-epoxidation reaction. This figure is used with permission from the American Chemistry Society[47]. NPs: Nanoparticles; TOF: turnover frequency; SERS: surface-enhanced Raman scattering.
Recently, Huang et al.[47] showed that the selective photo-epoxidation of a series of aliphatic and aromatic alkenes can be realized in liquid phase using Cu NPs supported on titanium nitride (TiN) by O2 or even air at moderate temperatures. The photocatalyst prepared by impregnation-reduction method exhibits well-dispersed metallic Cu NPs with a mean size of 4 nm supported on the TiN surface [Figure 3D], which shows excellent stability in photocatalytic styrene conversion. According to theoretical calculations, the Cu NPs remain in the metallic state under reaction conditions due to strong charge transfer between Cu NPs and the TiN support. Mechanistic study shows that the whole reaction takes place on the Cu surface. Firstly, the molecular oxygen reacts with cyclic ether solvent on the Cu surface under irradiation to produce oxygen adatoms (OA). Then, the photogenerated OA reacts with chemisorbed alkenes on Cu, forming a Cu-epoxide complex, and eventually desorbs as final products [Figure 3E]. Interestingly, they also observed product stereo-selectivity in trans-isomers for the epoxidation of stilbene.
The major advantage of photocatalytic epoxidation is the mild reaction conditions, which is ideal for some niche applications in medications and sterilizations that only require in situ generation of epoxides with relatively low concentration. This requires further investigations in robust, non-toxic photocatalyst materials, which can initiate the epoxidation of some light alkenes (i.e., ethylene and propene) under ambient conditions.
Oxidation of aromatics
Selective oxidation of aromatics to value-added products (i.e., alcohols or aldehydes) is important in the chemical industry as they are precursors for resins, electronics, and drugs[108,109]. The challenge in photocatalytic selective oxidation of aromatics is the development of photocatalysts with moderate oxidation power, which can partially oxidize specific functional groups while avoiding ring-opening and eventually CO2 formation. For example, photocatalytic direct oxidation of benzene to phenol has attracted great interest, as such a process may overcome the Hock rearrangement that produces an excessive amount of acetone as a byproduct. Chen et al.[110] showed that Fe-doped g-C3N4 is active in the oxidation of benzene to phenol under visible light. Although H2O2 is needed as the oxidant and the conversion is relatively low, the low cost of the photocatalyst material shows promising potential for applications. Yoshida et al.[111] showed that Pt-TiO2 can generate electrophilic oxygen species photocatalytically from water to initiate the oxidation of benzene to phenol under UV irradiation and deaerated conditions [Equation (5)]:

More importantly, a series of aromatics with different functional groups can be converted to corresponding phenolic compounds with relatively high selectivity. Mechanistic studies show that the reaction path is pH-dependent. Under neutral or acidic conditions, the photogenerated holes react with TiO2 to produce surface oxygen radicals, which attack the aromatic ring to form surface adsorbed phenoxy intermediates and eventually yield phenols. Under basic conditions, hydroxyl radical is the initial radical species that attacks the aromatic ring to produce phenols[112].
Researchers have further engineered the surface of photocatalysts with different strategies to improve the selectivity and conversion efficiency in benzene oxidation under ambient conditions using air or O2 as the oxidant. Zhang et al.[113] incorporated TiO2 NPs in hydrophobically modified mesocellular siliceous foam
Beyond the commonly used oxidants (i.e., O2 and H2O2), photo-oxidative conversion of aromatic compounds can also be performed with the presence of other oxidants. Jaynes and Hill[118] showed that selective carbonylation of toluene and cyclic alkanes to corresponding aldehydes can be achieved under 1 bar of CO by employing polyoxotungstate ([W10O32]3- or [PW12O40]3-), which is a type of polyoxometalates (POMs). During the reaction, the UV-excited photocatalyst first abstracts a hydrogen atom from the alkane to produce an alkyl radical, which is subsequently trapped by CO to yield an acyl radical. Then, the acyl radical is converted to aldehyde by reductive H• donation from the protonated photocatalyst. To further employ POMs under visible light region, Suzuki et al.[119] synthesized a sandwich-type Ce3+-containing silicotungstate (CePOM). An intramolecular charge transfer from Ce3+ to POM(W6+) is observed upon visible light excitation, which can initiate photocatalytic oxidative dehydrogenation of a series of primary and secondary aromatic amines in the presence of O2 (1 atm). Yamamoto et al.[120] showed that selectivity in oxidative coupling of benzene with cyclohexane is light sensitive using the Pd-modified TiO2 photocatalyst. Under UV irradiation, the photogenerated hole species react with benzene and cyclohexane to form corresponding radicals, thus resulting in non-selective formation of all possible coupling products (phenylcyclohexane, bicyclohexyl, and biphenyl). In contrast, high selectivity towards cross-coupling product (phenylcyclohexane) can be achieved under visible light irradiation due to a ligand-to-metal charge transfer of benzene complex adsorbed on TiO2, which produces benzene radical cation and selectively activates cyclohexane to form a cyclohexyl radical that attacks benzene molecule to form phenylcyclohexane. In addition, the Pd cocatalyst suppresses the formation of bicyclohexyl, thus resulting in a high selectivity towards the cross-coupling product. However, such a system shows a slow conversion rate due to poor light absorption under visible light.
Very recently, Cao et al.[121] reported a purely Bi-based semiconductor system (p-BWO nanosheets) for the efficient oxidation of toluene under visible light and ambient conditions (1 bar O2). As demonstrated in Figure 4A, the photocatalyst is constructed by crystalline Bi2WO6 (lattice spacings of 2.72 and 2.73 Å for the (002) and (200) planes, respectively) that is surrounded by amorphous BiOCl substance. Such structure provides numerous active sites at the crystalline/amorphous boundaries (orange and blue arrows). These amorphous BiOCl layers crystallize in situ under a continuous bombardment of high-energy electron beams [Figure 4B], indicating they are very sensitive to excited electrons. Owing to the unique microstructure for efficient charge separation, the lattice oxygen of the p-BWO nanosheets is very active upon visible light irradiation, which can be involved in the oxidation of toluene (leached out) and eventually healed by molecular oxygen. The p-BWO nanosheets show a 166-fold enhancement in photo-oxidation of toluene compared with that of pristine Bi2WO6 [Figure 4C].

Figure 4. (A,B) STEM images of p-BWO before and after electron beam bombardment. (C) Photo-oxidation rate of toluene for Bi2WO6 and p-BWO at different substrate loadings. (D) Photocatalytic cleavage of β-O-4 bond in lignin model compound (PP-ol). (E) Energy-band positions and photocatalytic performances of CdS with different sizes. (F) Photocatalytic conversion of PP-ol by several typical semiconductors. (G) Reusability of CdS NPs and CdS QDs under visible light irradiation. This figure is used with permission from Nature Publishing Group[121,122]. NPs: Nanoparticles; QDs: quantum dots.
Selective cracking of aromatic biomass by photocatalysis has also drawn significant attention very recently. Wu et al.[122] showed that CdS quantum dots (QDs) are efficient photocatalysts for the selective cracking of branched-chain of various lignin model compounds via β-O-4 bond cleavage under visible light irradiation and deaerated conditions, as demonstrated in Figure 4D. Meanwhile, due to the relatively weak oxidation power of CdS QDs, the functional groups and the aromatic ring structures remain stable during the photocatalytic process. The optimized CdS QDs photocatalyst with an average particle size of 4.4 nm presents a satisfactory conversion of lignin compound, which also exhibits significant enhancement compared with other commonly used photocatalysts [Figure 4E and F]. The CdS QDs photocatalyst also shows an enhanced performance compared with that of the CdS NPs counterpart in terms of both conversion rate and stability [Figure 4G]. More importantly, the CdS QDs photocatalyst is capable of converting native lignins and can be recycled via a facile aggregation-colloidization strategy, which shows huge potential for industrial applications.
Photocatalysis for selective oxidation of aromatics to value-added products has experienced a booming development. Further development in catalytic efficiency and selectivity may be achieved by further optimization of the adsorption of reactants and intermediates. The design of functionalized photocatalysts and cocatalysts may be inspired by a series of classic heterogeneous catalysis works, especially those surface science-related fundamental works. In addition, extensive efforts should be paid to tune the redox potential of the photocatalyst to avoid over-oxidation of specific functional groups and unwanted ring-opening side reactions.
Oxidative coupling reactions
Selective formation of bonds between carbon and other heteroatoms (i.e., N, S, and O) via cross-coupling is one of the important reactions in synthetic chemistry. The oxidative coupling reactions generally employ transition-metal catalysts to withdraw electrons from the reactants, which is accompanied by the addition of oxygen or deprotonation to the reactants. Employing metal-based molecules (i.e., Ru[(bpz)3](PF6)2 and Ru[bpy]3Cl2) as homogeneous photocatalysts has shown that a series of oxidative coupling reactions can be realized under visible light irradiation (i.e., phenol-arene coupling[123] and amines-enol silanes coupling[124]). To further enhance the efficiency and the reusability of the homogeneous dye complex, Tan et al.[125] immobilized the Ru[bpy]3Cl2 molecule into a radical MOF, FJI-Y2 for the photocatalytic cross-dehydrogenative coupling of N-phenyltetrahydroisoquinoline derivatives with phosphite esters to form α-aminoquinoline phosphonates. As demonstrated in Figure 5A and B, the [RuII(bpy)3] cations are encapsulated into the porous FJI-Y2 MOF via intermolecular interaction between pyridine rings of dye cations and phenyl rings of the MOF ligands. The FJI-Y2 MOF is characterized by multiple 1D channels with a window size of ~8 × 12 Å2, which offers an ideal platform with a large surface area and accessible active sites. It is also found that the dye-modified FJI-Y2 photocatalyst shows a higher performance than that of the homogeneous [RuII(bpy)3]Cl2 alone. The promotional effect originated from an efficient charge transfer due to intermolecular interactions between the 1,4,5,8-naphthalenediimide (NDI) group of the MOF and the RuII(bpy)3 catalyst, as demonstrated in Figure 5C. Under visible light irradiation, the NDI groups are excited to NDI*, which is immediately quenched by the excited RuII* via a single electron transfer (SET) process. Such process generates a RuI complex and a radical cation NDI+*, which separately react with an electron donor (here, D, N-phenyltetrahydroisoquinoline) and an oxygen [O] via SET process to form the corresponding radical cation (D+*) and superoxide radical [O]-*, respectively. Meanwhile, the NDI+* and RuI complex are relaxed to their ground states. The [O]-* further abstracts an H atom from the D+* to form the desired iminium ion, which undergoes nucleophilic addition to the desired product.

Figure 5. (A,B) [RuII(bpy)3] encapsulated in pores of FJI-Y2 MOF (highlighted as yellow atoms) and the interactions between the FJI-Y2 MOF ligands and [RuII(bpy)3] cations. (C) Cooperative catalysis mechanism of NDI radical and [RuII(bpy)3] photocatalyst via a SET pathway. This figure is used with permission from Elsevier[125]. (D) Atomic-resolution TEM image of R-WO3 nanosheets. (E) ESR spectra of D-WO3 and R-WO3. (F) Simulated differential charge density for O2 chemisorbed at a CUS W site of defective WO3. This figure is used with permission from the American Chemistry Society[126]. MOF: Metal-organic frameworks; NDI: naphthalenediimide; SET: single electron transfer; ESR: electron spin resonance.
The oxidative coupling reaction can also be realized by traditional metal oxide photocatalyst systems. Zhang et al.[126] synthesized defect-rich WO3 (R-WO3) nanosheets via calcination of hydrothermally prepared defect-deficient WO3 (D-WO3) in N2 atmosphere for aerobic coupling of primary amines to corresponding imines at room temperature [RT, Equation (6)]:

Such R-WO3 material contains numerous small pits on surface, creating a large surface area with more under-coordinated sites (CUS) for catalytic reactions. In high-resolution TEM imagery, lattice disorder and dislocation are observed in the R-WO3 nanosheets, revealing the presence of high-density defects [Figure 5D]. The massive defects result in a large number of electrons trapped at oxygen vacancies, as indicated by the significant signal of electron spin resonance (ESR) spectroscopy for R-WO3 (g = 2.002) compared with that of D-WO3 [Figure 5E]. Density functional theory (DFT) calculations indicate that molecular O2 is chemisorbed at the oxygen vacancy of D-WO3 surface via an end-on configuration, which is accompanied by a slight electron transfer from WO3 CUS site to O2 [Figure 5F]. The strong coupling between R-WO3 and O2 promotes oxygen reduction half reaction, thus accelerating the overall oxidative coupling.
Raza et al.[127] showed that the oxidative coupling of various amines to imines [Equation (6)] can also be realized by monolayer WS2 nanosheets under visible-light irradiation. The WS2 nanosheets synthesized via a modified liquid exfoliation method exhibit an average size of 120 nm with a height of 1 nm according to atomic force microscopy analysis, indicating most of the nanosheets are monolayer. The WS2 photocatalyst shows a similar reaction mechanism to that of the WO3 system; however, the monolayer WS2 nanosheets need to be operated under 50 °C. Xu and Fu[128] also developed core-shell structured sulfide-g-C3N4 to realize oxidative coupling of amines. A 4 nm thick shell of g-C3N4shell is coated on the CdScore nanowire via a self-assembly process, which constructs a heterojunction interface for enhanced charge separation of photogenerated electrons and holes. Nevertheless, it has also been noticed that a much faster conversion rate can be achieved by using H2O2 as the oxidant, therefore suggesting that the formation of •O2- radical is the rate-determining step for the photocatalytic oxidative coupling of various amines.
Our group recently reported that the identity of metal NP-decorated g-C3N4 is essential in controlling the selectively photocatalytic dehydrogenative homocoupling of primary amines into imines and secondary amines[129]. While Pt NPs drive the dehydrogenative homocoupling of primary amines into corresponding imines, Pd NPs promote self-hydrogenation of the photogenerated imines into secondary amines using the H atoms abstracted from the amines [Figure 6A]. Mechanism studies reveal that the adsorption of photogenerated imine molecule is relatively strong on Pd compared to on Pt NPs according to temperature programmed desorption (TPD, Figure 6B), thus facilitating the hydrogenation of imine into secondary amine. Moreover, a significant evolution of molecular hydrogen is observed for Pt/C3N4 under irradiation when primary amine and secondary amine are present, whereas no detectable H2 is spotted for Pd/C3N4 [Figure 6C and D]. Moreover, a distinct delay of H2 evolution is observed on Pd/C3N4 (~3 h after immense irradiation, red curve) in the photocatalytic hydrogenation of imine (2a) using isopropanol as the hydrogen donor [Figure 6E]. This further confirms that the abstracted hydrogen atoms from the amine molecules can be temporarily stored on Pd NPs, thus hydrogenating imine molecules into secondary amine without additional hydrogen donors (i.e., alcohols).

Figure 6. (A) Selective photocatalytic conversion of primary amines tuned by metal cocatalyst and the reaction pathways. (B) TPD analysis of imine adsorption on Pt/C3N4 and Pd/C3N4. (C,D) In situ MS analysis of H2 evolution during photocatalytic conversion of amine and secondary amine on Pt/C3N4 and Pd/C3N4 under deaerated conditions. (E) H2 evolution during photohydrogenation of 2a in isopropanol. The figures are used with permission from the American Chemistry Society[129]. TPD: Temperature-programmed desorption; MS: mass spectrometry.
Very recently, the dehydrogenative coupling of simple aliphatic alcohol (methanol) to the value-added product [ethylene glycol (EG)] was realized by Xie et al.[130] using the metal sulfide composite. While conventional photocatalysts (i.e., TiO2, g-C3N4, and Cu2O) only oxidize methanol into formaldehyde, CO, and CO2, CdS NPs and nanorods produce a reasonable amount of EG. Interestingly, both the conversion rate of methanol and the selectivity to EG can be significantly enhanced by loading MoS2 nanofoam cocatalyst onto the CdS nanorod [Figure 7A and B]. ESR analysis reveals that mainly hydroxymethyl radicals (•CH2OH) instead of methoxy radicals (CH3O•) are generated under irradiation using CdS photocatalyst [Figure 7C], suggesting a preferential activation of C-H bond rather than O-H bond of methanol by photoexcited holes. Eventually, the photogenerated •CH2OH radicals readily desorb from catalyst surfaces for subsequent coupling. This work presents unique photocatalytic C-H activation that may offer an alternative process for cheap aliphatic alcohol conversion in the petroleum industry.

Figure 7. (A,B) TEM images of MoS2 foam/CdS. (C) In situ ESR spectra of MoS2 foam/CdS catalyst in methanol-DMPO solution with or without irradiation. This figure is used with permission from a Creative Commons Attribution License[130]. ESR: Electron spin resonance.
Nevertheless, plasmonic Au-Pd alloy NPs supported on ZrO2 have also been employed as photocatalyst for a series of C-C coupling reactions (i.e., Sonogashira coupling, Stille coupling, and Hiyama coupling) with high yields[131]. It is proposed that the alloy NPs absorb visible light and transfer excited electrons to the Pd sites to initiate the reactions. Beyond conventional metal-based semiconductor and plasmonic metal NPs photocatalyst systems, Ghasimi et al.[132] demonstrated that the conjugated organic polymer can be used as a Pd-free photocatalyst for efficient Stille coupling reaction to construct aromatic C-C bond under visible light irradiation. The photocatalyst polymer, namely P-Az-B, can be obtained via Suzuki cross-coupling reaction of 1,3-dibromoazulene with 1,3,5-phenyltriboronic acid tris(pinacol) ester, where the 1,3-dibromoazulene is a co-monomer to obtain high porosity of the material. The P-Az-B photocatalyst exhibits excellent light absorption with an optical band gap of 2.03 eV. In addition, the highest occupied molecular orbital and the lowest unoccupied molecular orbital (LUMO) of P-Az-B (+1.14 and -1.10 V vs. saturated calomel electrode) are comparable to that of the Ru-based complex, suggesting its compatibility for driving C-C coupling reactions. Upon visible light irradiation, the photogenerated hole and electron react with aryl stannane and the aryl iodide, resulting in the formation of destannylation radical and aryl halide radical, respectively. Then, the radicals desorb from the photocatalyst and react to create a substitutional intermediate, which finally leads to the formation of C-C bond by releasing the iodide anion.
The cross-coupling of aniline and alcohol for the synthesis of asymmetric secondary amines can also be realized by heterogeneous photocatalytic systems. Shiraishi et al.[133] demonstrated that the Pd/TiO2 photocatalyst promotes the N-monoalkylation of amine with alcohol via tandem photocatalytic and catalytic reactions: (i) photo-oxidation of alcohol on Pd/TiO2; (ii) condensation of photogenerated aldehyde with amine on TiO2; and (iii) hydrogenation of the produced imine by the photogenerated surface H atoms on the Pd particles from alcohol oxidation. The rate-determining step is the imine hydrogenation, which strongly depends on the number of surface Pd atoms on the triangle site of Pd particles [Figure 8A]. Meanwhile, the strong adsorption of alcohols on larger triangle sites of Pd NPs should be avoided as it will suppress the imine hydrogenation. Therefore, Pd NPs with an optimum size of 2-2.5 nm contain a relatively larger number of triangle site Pd atoms but do not show strong alcohol adsorption, present the highest activity for imine hydrogenation and promote N-monoalkylation of amine with alcohol [Figure 8B]. Similarly, the rapid photocatalytic N-alkylation of pharmaceutically relevant amines with alcohols can also be realized by employing a mixed heterogeneous Cu-Au/TiO2 photocatalyst[134]. By carefully controlling the reaction conditions, selective mono- and di-alkylation of primary amines and the non-symmetrical dialkylation of primary amines to hetero-substituted tertiary amines can be achieved. In this scenario, alcohol is typically used as the solvent, which serves as both the reactant to couple with amine and a hydrogen donor to supply H atoms for the hydrogenation of the photogenerated imines. Therefore, selectivity control over imine and secondary amine and the formation of aldehyde byproducts remain challenges.

Figure 8. (A) The fcc cuboctahedron model of Pd particles. (B) Relationship between the Pd particle size and the turnover number for hydrogenation of imine. This figure is used with permission from the American Chemistry Society[133]. (C) Metal cocatalyst tuned photocatalytic cross-coupling of aniline and alcohol. (D) TPD analysis of benzyl alcohol adsorption on Rh/TiO2 and Fe/TiO2, respectively. (E) ESR spectra of Rh/TiO2 and Fe/TiO2 in DMSO under irradiation. This figure is used with permission from Elsevier[135]. TPD: Temperature programmed desorption; ESR: electron spin resonance; DMSO: dimethyl sulfoxide.
A very recent work provides an alternative strategy for selective cross-coupling of amines and alcohols without the use of additional hydrogen donors[135]. This is realized by controlling the identity of the metal cocatalysts supported on TiO2 to mediate the photocatalytic activation of alcohols, thus separating the stepwise dehydrogenative-condensation process to imine (Rh) and the direct condensation process to secondary amine (Fe, Figure 8C). TPD and ESR analysis reveals that Rh exhibits strong adsorption of alcohol with fast generation kinetics of the •OH radicals [Figures 8D and E], thus leading to a rapid photo-oxidation of alcohol into aldehyde and the consecutive condensation of aldehyde with aniline. In contrast, the Fe/TiO2 displays a weak interaction with alcohol and slow kinetics in •OH formation, resulting in a mild activation of the alcohol for direct condensation with aniline to yield asymmetric secondary amine.
Up to date, the homo-coupling of some model compounds by photocatalysis has been well established and highly efficient photocatalysts have been developed. Further investigations in cross-coupling reactions are crucial for applications that require careful control over the active sites of the catalyst and reaction conditions. This calls for an in-depth understanding of reaction pathways, intermediates, and radical species. One could learn from traditional homogeneous catalysis of the reaction mechanisms to help the design of heterogeneous photocatalyst. Additionally, the quantification of unwanted byproducts and unknown products should not be neglected, as this may give some useful hints for the design of highly selective photocatalysts.
NANOSIZED PHOTOCATALYSTS FOR REDUCTION REACTIONS
Hydrogenation of CO2 and CO
Photocatalytic CO2 reduction is a very hot topic in photocatalysis and photo-electrocatalysis. Various photocatalysts ranging from simple oxides, sulfides, perovskites, MOFs[136], and plasmonic metal NPs[137] to composites[138] have been developed for CO2 reduction with or without sacrificial electron donors. The design of cocatalyst has also been extensively investigated to further boost the photocatalytic CO2 reduction performance[139-141]. Since there are a number of in-depth reviews on this topic[136,142], we focus on the hydrogenation of CO2 and CO by molecular H2 and the incorporation of CO2 into other reactants for the formation of more complicated chemicals.
Sastre et al.[143] showed that the p-type NiO photocatalyst can reduce CO to CH4 with the presence of water under visible light irradiation at RT. It is also noticed that CO reduction to CH4 can be significantly enhanced with the presence of H2 as a reducing agent. In addition, the deposition of elemental carbon (coking) on the photocatalyst surface is also observed. It is therefore supposed that the NiO photocatalyst not only accelerates the CO reduction by water [Equation (7)] but also promotes a Fischer-Tropsch-like process following Equation (8):
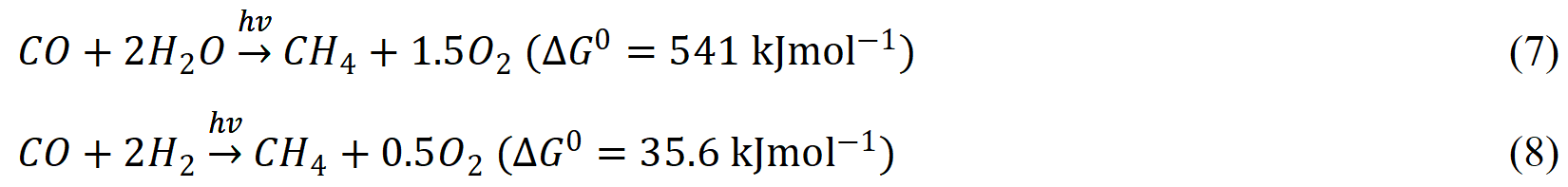
Based on these findings, the same group further explored the photocatalytic Fischer-Tropsch process directly from CO2 and H2[144]. It has been found that, although NiO exhibits high conversion of CO2 to CH4, the photocatalyst gradually deactivates upon reuse. Meanwhile, nickel supported on porous silica-alumina (Ni/SixAlyOz) is an efficient and reusable photocatalyst for the reduction of CO2 with comparable performance. As demonstrated in Figure 9A, it is proposed that the reaction is initiated via H2 activation by the photogenerated charge carriers, where Ni-H species are generated to further activate CO2 to form surface adsorbed, hydrogenated intermediates and eventually release as CH4. Since the silica-alumina is inert support, the reaction is more likely a photo-thermal catalytic process that is driven by the localized hot electrons generated from plasmonic absorption of Ni metal NPs. Researchers should be aware that the Ni/SixAlyOz photocatalyst only produces CH4 and CO without the formation of hydrocarbons with longer chains. To further promote the photocatalytic Fischer-Tropsch process, Guo et al.[145] deposited worm-like Ru nanostructures on graphene as photocatalyst for the conversion of CO to more complicated hydrocarbons under H2 atmospheres. Although the Ru/graphene photocatalyst shows a decent selectivity to liquid fuel products under visible light irradiation (C5+, > 70%), such a system requires a relatively high operation temperature (150 °C), and the activity is more dependent on heat rather than light.

Figure 9. (A) Complete photocatalytic CO2 hydrogenation with H2 using Ni supported on silica-alumina. This figure is used with permission from the American Chemistry Society[144]. (B,C) EDS line-scan and HRTEM image of the CoFe alloy NPs used for photothermal CO2 hydrogenation to hydrocarbons with H2. (D) Temperature profiles during photothermal CO2 hydrogenation. This figure is used with permission from Wiley-VCH[146].
Very recently, Zhang’s group showed that the synthesis of hydrocarbons from syngas (CO and H2) can be realized by photo-thermal catalytic approach at RT[58,146-149]. This type of nanostructured photocatalysts is generally derived from hydrogen reduction of layered double hydroxides (LDH, i.e., ZnCoAl, ZnFeAl, and CoFeAl) at elevated temperatures (300 °C -700 °C), yielding metal/metal oxide composites (i.e., Co/Co3O4, Fe/FeOx, and CoFe alloy) leached out from the LDH and naturally loaded on the corresponding inert support (ZnO, Al2O3). The activity and selectivity of these LDH-derived photocatalysts can be tuned by optimizing the annealing temperature and identity of the metal species in the LDH. While the photocatalyst derived from ZnCoAl LDH shows a reasonable CO conversion (15.4%) and selectivity towards light olefin (C2-C4=, ~19%), the performance can be further enhanced by employing ZnFeAl LDH as the photocatalyst precursor (Con. > 20%, Sel. > 40%) due to a suppression of CO2 formation. Furthermore, direct hydrogenation of CO2 can also be realized by fabricating the CoFe alloy NPs from annealing of CoFeAl LDH nanosheets under H2 atmosphere, as shown in Figure 9B and C. The CoFe alloy NPs synthesized at 650 °C show remarkable selectivity toward hydrocarbons (60% CH4, 35% C2+). It should be clear that the catalytic activity of these photocatalysts originates from the photothermal effect, as the temperature of the catalyst bed increases rapidly upon irradiation with the presence of photocatalyst [Figure 9D].
For carboxylation with CO2, Ye et al.[150] showed that radical chemistry possesses great potential to bring CO2 to a new stage using either a transition-metal catalyst or a photocatalyst. Nevertheless, incorporation of CO2 reduction with oxidation of other reactants to synthesize valuable chemicals has also emerged recently. Yang et al.[151] showed that the photocatalytic reduction of CO2 can be coupled with the oxidation of amine by using Cu/TiO2 photocatalyst. While CO2 is reduced to CH3OH by photo-induced electrons, benzylamine reacts with photogenerated holes and undergoes dehydrogenation coupling to produce imine. Such a system provides an economic strategy to fully utilize the photogenerated charge carriers. Chen et al.[152] reported the utilization of Cu2O/Cu nanocomposite for photocatalytic coupling of CO2 with benzyl alcohol. Under visible light irradiation, benzyl acetate rather than C1 products is selectively produced over Cu2O/Cu, as described in Equation (9). Upon irradiation, the photogenerated holes react with benzyl alcohol to form benzaldehyde with the release of two protons. The protons along with photogenerated electrons interact with surface-adsorbed CO2 to generate CH2O via hydrogenation of the surface-adsorbed CO* intermediate, during which CO2 undergoes a facile cleavage of the C-O bond on Cu sites. CH2O further couples with a neighboring CO* to form surface adsorbed H2C-CO. The H2C-CO intermediate converts to surface-adsorbed H3C-CO via H abstraction from benzyl alcohol and subsequently undergoes a coupling reaction with the benzyl oxygen ion to produce benzyl acetate. However, researchers should be aware that the Cu2O/Cu material transforms to Cu2O/CuO after photocatalytic reaction, which needs to be regenerated by NaBH4.

Photocatalytic CO2 reduction is a challenging and intriguing reaction; however, the value of the products and the inherent slow reaction kinetics render its future questionable from an economical viewpoint. Nevertheless, fundamental investigations are still important in this field, but they should be focused on reaction mechanisms rather than reporting performance. It is also important to use standard procedures for performance testing and reporting.
Hydrogenation of O2
H2O2 is widely used in many applications including medication, bleaching, and production of organic chemicals. Previously, industrial synthesis of H2O2 was realized by hydrolysis of ammonium peroxydisulfate ([NH4]2S2O8) that is obtained by electrolysis of ammonium bisulfate (NH4HSO4) in sulfuric acid. Nowadays, H2O2 is mainly manufactured by the anthraquinone process, as demonstrated in Equation (10)[153]:
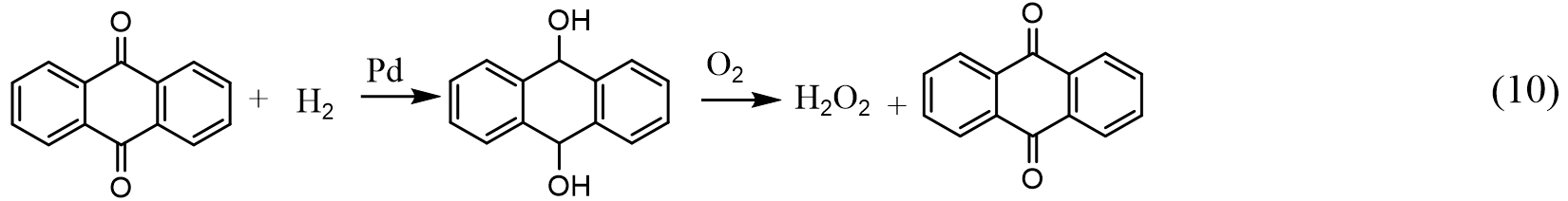
where the anthraquinone (i.e., 2-ethylanthraquinone) is hydrogenated to the corresponding anthrahydroquinone and consequently oxidizes back to anthraquinone and produces H2O2. The economics of this process depends on the Pd-based hydrogenation catalyst, the effective recycling of the extraction solvents, and the cost of quinones.
Since it has been proven that reactive oxygen species including O2•-, OH•, and H2O2 are produced during the photocatalytic decomposition of organic pollutants, it is reasonable to consider the photosynthesis of H2O2 by employing a designed semiconductor photocatalyst under mild conditions with appropriate hydrogen sources. Teranishi et al.[154] showed that H2O2 (10 mM level) can be synthesized by simply using Au NP-modified TiO2 with ethanol as the hydrogen source under aerobic conditions. Kinetic analysis reveals that, while Au NPs on TiO2 promote the formation of H2O2 compared with that of pristine TiO2 or Pt/TiO2, the decomposition rate of H2O2 on Au/TiO2 is also significantly suppressed. It is therefore proposed that the hydrogenation of O2 and the degradation of H2O2 take place on the metal surface and the TiO2 surface, respectively, and surface engineering of the photocatalyst material is the key to further optimize the synthesis of H2O2. The photocatalytic H2O2 production over Au/TiO2 can be further enhanced by adding borate or phosphate in alkaline water solution[155], which is ascribed to an anion-mediated hole transfer from Au/TiO2 to electron donor (methanol). In addition to the anion effect, Moon et al.[156] showed that the metal promoter on TiO2 can be replaced by reduced graphene oxide (rGO) for the photocatalytic production of H2O2. Interestingly, by adding cobalt salt to the phosphate-containing catalyst suspension, cobalt phosphate complex (CoPi) that is often served as an electrocatalyst can be formed in situ on the rGO/TiO2 surface. The loading of CoPi as cocatalyst further reduces the overpotential of water oxidation, therefore enabling photocatalytic production of H2O2 even without organic hydrogen resources (i.e., alcohols).
More efforts have been devoted to realizing the selective synthesis of H2O2 under visible light conditions. Shiraishi et al.[157] employed g-C3N4 as the visible light response photocatalyst to generate H2O2 selectively (> 90%) with the presence of alcohol and O2. Mechanistic studies reveal that the high selectivity to H2O2 originates from the efficient formation of surface-adsorbed 1,4-endoperoxide species (C-O-O-N), which suppresses the one-electron reduction path of O2 to superoxide radicals. He et al.[158] showed that the dimension of carbon support materials is crucial in tuning the recombination rate of charge carriers and light absorption properties, which is essential for boosting the evolution rate of H2O2. Li et al.[159] demonstrated that the performance of g-C3N4 can be further improved by creating carbon vacancies, which reduce the bandgap and promote the delocalization of excited electrons of the material. The carbon vacancies not only provide more sites for interfacial charge transfer from the photocatalyst to adsorbed O2, but they also alternate the H2O2 generation pathway from a two-step single-electron process to a one-step two-electron direct reduction.
Kim et al.[160] further explored the possibility of coupling industrial H2O2 synthetic process with photocatalysis. As demonstrated in Figure 10A, the anthraquinone molecule [anthraquinone-2-carboxylic acid (AQ)] is anchored onto g-C3N4 via a catalyst-free, direct dehydration coupling of the -COOH of AQ and the -NH2 of g-C3N4. Upon visible light excitation, isopropanol is oxidized on the g-C3N4 surface to acetone and releases protons. Meanwhile, the photogenerated electrons and abstracted protons transfer to the anchored AQ molecule, reducing it to surface-anchored anthrahydroquinone (AQH2). Eventually, the anchored AQH2 is oxidized back to AQ in the presence of O2, resulting in the production of H2O2. The efficient anchoring of AQ has been confirmed by X-ray photoelectron spectroscopy (XPS, Figure 10B). While the C1s peaks of g-C3N4 are assigned to the adventitious carbon and defect-derived sp2-carbon (284.8 eV) and the N-C = N bond in the triazine units (288.2 eV), a strong aromatic C-C bond (284.8 eV) is observed when AQ is coupled with g-C3N4. In addition, Fourier transform infrared spectroscopy (FT-IR) has also spotted increases in absorption at 1278 and 1676 cm-1, which correspond to C-N and C = O stretching vibrations due to the formation of amide bond. Owing to the efficient interfacial charge transfer and the superior activity of AQ in O2 reduction, such an anchored system exhibits a high apparent quantum yield in H2O2 production of 19.5% at 380 nm, which is significantly higher than that of pristine g-C3N4 or g-C3N4 modified with metallic co-catalysts.

Figure 10. (A) g-C3N4 coupled with AQ(-COOH) for photocatalytic hydrogen peroxide generation under solar light irradiation. (B) XPS spectra of C1s on AQ(-COOH), g-C3N4, and g-C3N4/AQ(-COOH). This figure is used with permission from Elsevier[160]. (C) Solar-to-chemical conversion efficiency of PCN, PCN_Na15, and Sb-SAPC15 under AM 1.5 illumination for H2O2 production. (D) TEM image of the Sb-SAPC15 (Scale bar, 2 nm). (E) Raman spectra recorded during photoreaction in isopropanol solution with saturated oxygen. This figure is used with permission from Nature Research[161]. XPS: X-ray photoelectron spectroscopy.
Very recently, Teng et al.[161] showed that a single Sb atom dispersed on g-C3N4 (Sb-SAPC) can achieve a remarkable apparent quantum yield of 17.6% for H2O2 synthesis under 420 nm irradiation. The solar-to-chemical conversion efficiency of Sb-SAPC15 reaches 0.61%, which is higher than most water-splitting photocatalysts [Figure 10C]. TEM imaging shows that high-density Sb atoms are uniformly dispersed over the entire carbon nitride matrix [Figure 10D]. Raman spectroscopy has been performed to identify the intermediate in the photocatalytic process with the presence of isopropanol as the electron donor [Figure 10E]. A new absorption at 855 cm-1 is observed for the Sb-SAPCs, which can be assigned to the O-O stretching of an Sb-OOH species that corresponds to the end-on adsorption configuration of molecular oxygen. Such end-on adsorption notably suppresses the 4e- oxygen reduction reaction, thus leading to a high selectivity of the 2e- process for H2O2 synthesis.
Photocatalysis provides an alternative approach for the on-site synthesis of H2O2, which is ideal for coupling with some specialized fine chemical syntheses that require a relatively low concentration of H2O2. This requires leveling up the concentration of photogenerated H2O2 from ppm level to sub wt% level. One possible solution is coupling the oxygen reduction with water oxidation to produce H2O2 via both cathodic and anodic reactions using designed photocatalysts. In addition, the degradation kinetics of photogenerated H2O2 is worthy of investigation. It is also necessary to evaluate the poisoning effect of H2O2 at elevated concentrations for the optimization of photocatalysts.
Hydrogenation of organic molecules
Early investigations showed that fine TiO2 NPs and ZnS quantum dots photocatalyst can be used to hydrogenate a series of organic molecules (i.e., propene, aromatic ketones, and amines) upon UV excitation[162-164], although the light utilization, quantum efficiency, conversion, and selectivity are far from satisfactory in general. To overcome these challenges, many efforts have been focused on surface engineering of photocatalyst materials. Hao et al.[164] reported surface engineered heterogeneous photocatalysts for the hydrogenative fixation of N2 to NH3. Zhang et al.[165] showed that functionalization of TiO2 with metal NPs can significantly promote the hydrogenative cyclization of levulinic acid (LA) into γ-valerolactone (GVL, a potential green fuel) selectively using isopropanol as the hydrogen source, as demonstrated in Equation (11):

Here, isopropanol is dehydrogenated to acetone as the main product, donating a proton to LA that is cyclized to form GVL via the formation of acetyl propionyl radical. It is observed that while the polymorph composition of TiO2 influences the conversion rate of LA, the identity of metal NPs controls the selectivity. Au NPs exhibit the highest LA conversion (79%) and GVL selectivity (85%) compared with that of Pd and Pt promoters, which is compatible with thermo-catalytic hydrogenation approach using Cu/ZrO2 and Ru/C catalysts.
Lots of efforts have also been made to initiate hydrogenation reactions upon visible light radiation. Kohtani et al.[166] showed that, by anchoring organic dye molecules on pristine TiO2 NPs, hydrogenation of selected aromatic ketones to corresponding alcohols with the presence of a hydrogen donor (trimethylamine) can be achieved under visible light excitation (> 400 nm). Although the utilization of metal-free dyes (fluorescein and rhodamine B) is an economical and feasible solution, the poor stability limits its applications. Jiao et al.[167] developed a Pd supported on β-SiC photocatalyst for the hydrogenation of furan molecules under visible light excitation at RT [Equation (12)]. Although β-SiC is a narrow bandgap semiconductor (~2.4 eV), it is found that loading of Pd NPs (3 wt%) is essential to initiate the efficient hydrogenation of furan and its derivatives. The authors proposed that a Mott-Schottky contact of SiC and Pd enhances the charge transfer of photogenerated electrons from SiC to Pd NPs, thus promoting the catalytic activity; however, the photo-thermal catalytic approach induced by the plasmonic absorption of Pd NPs cannot be ruled out.
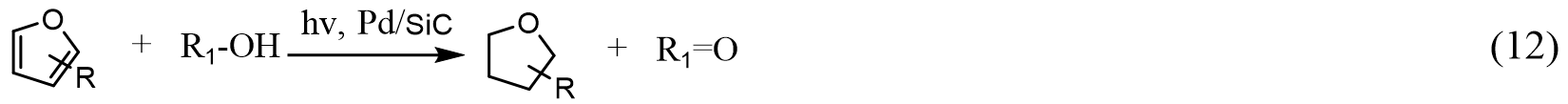
The same group further employed SiC with Au NPs for the selective hydrogenation of cinnamaldehyde using isopropanol as a hydrogen source under deaerated conditions [Equation (13)][168]:

Figure 11A presents that Au NPs with a particle size ranging from 3-8 nm are evenly deposited on the SiC support. Remarkably, the Au/SiC system exhibits excellent selectivity and reasonable conversion in the whole visible light range, as demonstrated in Figure 11B. A very high turnover frequency (TOF, 487 h-1) can be achieved at optimum conditions (30 mg catalyst and 20 mg KOH in 10 mL isopropanol and Xe‐lamp with light intensity of 1.0 W•cm-2). Nevertheless, the Au/SiC photocatalyst is capable of hydrogenating a series of α,β‐unsaturated aldehydes to their corresponding unsaturated alcohols with high conversion and selectivity. It should also be noted that, in contrast to the hydrogenation of furans by Pd/SiC, the hydrogenation of cinnamaldehyde by Au/SiC is definitely a photo-thermal catalytic process that is induced by the plasmonic absorption of Au NPs. While the oxidation of 2‐propanol takes place on the positively charged Au NPs surface, α,β‐unsaturated aldehydes are adsorbed on the Au/SiC interface and reduced by the photogenerated electrons to unsaturated alcohols.

Figure 11. (A) TEM image of the 1 wt% Au/SiC photocatalyst and size distribution of Au NPs. (B) Selective photocatalytic hydrogenation of cinnamaldehyde by Au/SiC at different radiation wavelengths. (C) Asymmetric and symmetric adsorption of nitrobenzene on rutile TiO2 (110) surface. Light blue and green balls are Ob atoms, while red and yellow balls are Ti and H atoms. (D) DRIFT spectra of nitrobenzene adsorbed on selected anatase (A) and rutile (R) TiO2 at 303 K. This figure is used with permission from the American Chemistry Society[168,169]. NPs: Nanoparticles; DRIFT: diffuse reflectance infrared Fourier transform.
Shiraishi et al.[169] showed that the photocatalytic selective hydrogenation of a series of nitroaromatics to anilines with isopropanol as the hydrogen donor is sensitive to the polymorph composition of TiO2 [Equation (14)]:
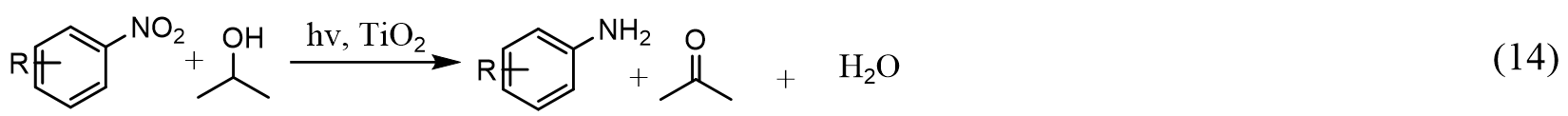
Interestingly, while anatase and P25 TiO2 show poor activity and selectivity towards aniline, photoactivated rutile TiO2 samples all show high performance in selective hydrogenation of nitroaromatics. It is proposed that the adsorption of nitroaromatics on rutile is responsible for the enhanced performance. As demonstrated in Figure 11C, the adsorption and hydrogenation of nitroaromatics on the photoexcited TiO2 surface take place via either asymmetric adsorption on hydroxyl (TiOH) sites or symmetric adsorption on bridging oxygen vacancy (Ob vac.) sites. While the symmetric adsorption site can donate two photogenerated electrons to a nitroaromatic molecule, the asymmetric adsorption site only provides one electron at once, thus resulting in a poor performance in reduction. Diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy has confirmed a significant density of symmetric adsorption sites on different rutile TiO2 compared with that of anatase samples [Figure 11D], therefore leading to a fast aniline formation rate with high quantum yields (> 25%).
New materials have also been developed to drive the hydrogenation of nitroarenes under visible light conditions. Toyao et al.[170] synthesized an amino-functionalized Ti(IV)-MOF modified with Pt NPs (Pt/Ti-MOF-NH2) for this purpose with reasonable stability. In situ ESR analysis reveals that the excited electrons on the organic linker transfer to the deposited Pt NPs via the titanium-oxo clusters, which promote the spatial charge transfer, thus resulting in enhanced performance. At the optimized Pt loading (1.5 wt%), Pt/Ti-MOF-NH2 shows enhanced performance in both H2 production and nitrobenzene reduction. However, low quantum efficiencies are observed for this system (~1%-2% under visible light irradiation), probably due to the rapid evolution of molecular H2 that competes with the hydrogenation of nitrobenzene. Gao et al.[171] employed nanosized Ni2P as the cocatalyst to modify CdS as a photocatalyst to realize the hydrogenation of a series of nitroarenes with Na2S/Na2SO3 as sacrificial electron donors. In this system, water donates a proton to hydrogenate nitroarenes, as the Na2S/Na2SO3 contains no proton. It is also observed that the photocatalytic H2 evolution competes with the photosynthesis of aniline on the Ni2P surface. Therefore, further surface engineering of the photocatalyst to suppress the H2 evolution is essential to enhance the reduction of nitroarenes.
Very recently, Xiao et al.[172] demonstrated that chemoselective hydrogenation of various nitroarenes to corresponding anilines can be achieved by simply using the pristine g-C3N4 photocatalyst under visible light radiation. The process shows excellent yields (82%-100%) and is even applicable to gram-scale reactions. However, it should be noted that such a process is operated at elevated temperature (70 °C-90 °C) and requires the addition of hydrazine hydrate (N2H4·H2O) as the hydrogen source and hole scavenger (strong reducing agent with very high toxicity), which hinders its application in large scale due to safety issues. Nevertheless, Huang et al.[173] showed that Au supported on ZrO2 photocatalyst can initiate the hydrogenation of nitroaromatics under visible light with formic acid and water as the hydrogen source. Since ZrO2 is a large bandgap semiconductor (> 5 eV), the activity in visible light spectrum originates from the plasmonic excitation of Au NPs. In addition, Au/ZrO2 is a versatile photocatalyst for the selective hydrogenation of various unsaturated bonds (i.e., C=C, C≡C, C=O, and C=N) under mild reaction conditions. Theoretical calculations reveal that direct photoexcitation of hybridized orbitals that are generated from chemisorbed reactants and metals is the driving force of the hydrogenation reaction.
Heterogeneous photocatalysis provides an ambient platform for hydrogenation reactions that employs hydrogen-containing molecules instead of high-pressure H2. Such a mild reaction condition may also benefit semi-hydrogenation of alkynes and selective hydrogenation of complicated pharmaceutical molecules with vulnerable functional groups. In any case, the utilization of abundant and safe hydrogen donors (i.e., alcohols, ammonia) is recommended rather than the use of toxic, expensive, and risky hydrogen sources (e.g., hydrazine). The design of cocatalysts with tunable hydrogen adsorption energy is also vital for efficient and selective hydrogenation, which may be promoted by the experience of heterogeneous hydrogenation investigations.
Reductive coupling reactions
Reductive coupling reactions (i.e., C-C, C-N, N=N) via homogeneous catalysis have been well investigated[174-184], especially in transition metal-catalyzed cross-coupling reactions promoted under visible light[183]. However, the need for expensive catalysts, catalyst separation, harsh reaction conditions, and toxic reducing agents (i.e., PH3, Tl, and NaTeH) limit their applications. Therefore, there is a big demand to develop heterogeneous photocatalyst systems that can realize coupling reactions selectively under ambient conditions. Zhang et al.[184] demonstrated the effectiveness of semi-heterogeneous metallaphotocatalysis using polymeric carbon nitride photocatalyst for C-N cross-coupling reactions. Moreover, Wang et al.[185] showed that, by constructing unique plasmonic Au-Pd nanostructures, Suzuki coupling reaction of a series of aromatic compounds can be realized via harvesting visible-to-near-infrared light [Equation (15)]:

Here, the Au-Pd nanostructures consist of Au nanorods for light harvesting via plasmonic excitation, whereas the supported Pd NPs, through seeded growth, serve as catalytic active sites that are frequently used in conventional Suzuki coupling reactions. In addition, cetyltrimethylammonium bromide is required to bring bromobenzene into the aqueous reaction solution under basic conditions. Such a system shows a rapid temperature rise under infrared illumination, revealing a photo-thermal catalytic behavior that originated from the oscillation of localized hot electrons. The higher efficiency observed for the photo-induced coupling compared with that of the thermally induced process indicates that the local temperature may be significantly higher on the plasmonic catalyst surface than in the solution. More importantly, the Au-Pd nanostructures have been further tailored to directly harvest sunlight to drive coupling reactions, which show industrial potential for fine chemical synthesis. This design concept of Au-Pd was also employed by Xiao and coauthors for various C-C coupling reactions[131]. However, the conversion rate in terms of turnover frequency, selectivity, and quantum efficiency still needs to be enhanced for applications. Lanterna et al.[186] later showed that the plasmonic Au NPs can be supported on TiO2 to realize visible light-induced reductive C-C coupling of a variety of substituted benzyl bromides [Equation (16)]:
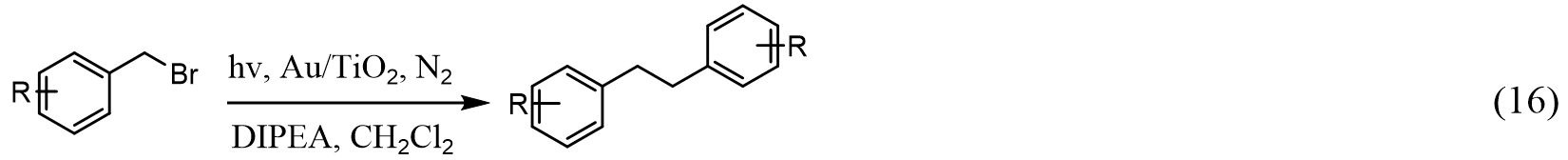
Here, the reductive coupling of benzyl bromides requires careful selection of both electron donors and solvents, where the combination of diisopropylethylamine and CH2Cl2 yields the optimum photocatalytic performance. Although the neat Au/TiO2 photocatalyst system shows decent conversion and selectivity to desired products in general, further engineering of the material is needed to improve the stability.
Li and coworkers recently depicted the rational design of metal cocatalyst for photocatalytic dehalogenative coupling of benzyl bromide based on the Sabatier principle[187]. The optimal activity and selectivity require appropriate adsorption energies of the intermediates (benzyl radical, bromine atom, and surface adsorbed hydrogen atoms (Hads)) on the metal cocatalyst. DFT calculations predict that Cu is the promising candidate for this reaction among a series of transition metals owing to the optimized adsorption of benzyl radical and Br atom [Figure 12A]. This prediction is confirmed experimentally, where Cu indeed presents high conversion and selectivity in homocoupling of benzyl bromide without the formation of toluene [Figure 12B]. Remarkably, this design strategy can be generalized for the modification of other photocatalysts. The Cu deposited on g-C3N4 (Cu/C3N4) presents a decent performance towards the conversion of benzyl bromide into bibenzyl under visible light irradiation.

Figure 12. (A) ΔE(ArCH2•) as a function of ΔE(Br) for the screening of metal cocatalysts for photocatalytic dehalogenative coupling of benzyl bromide (the numbers in parentheses are ΔE(Hads) of the corresponding metals). (B) Photocatalytic benzyl bromide coupling using a series of selected metals supported on TiO2. This figure is used with permission from the American Chemistry Society[187]. (C) Picture of molecular adsorption on CNs. (D) T1/T2 ratio of different moieties of perfluorohexyl iodide in various CN-based materials. This figure is used with permission from American Association for the Advancement of Science[188].
Filippini et al.[188] reported the amorphous g-C3N4 (am-CN) material for visible light-driven C-C bond formation towards the synthesis of valuable perfluoroalkylated intermediates from the corresponding iodides. The binding of perfluorobutyl iodide (C4F9I) occurs via halogen bonding with the N atom of the am-CN. This step affects the rate of the halogen dissociation of C4F9I to form the ∙C4F9 radical [Figure 12C]. The affinity of reagents toward the solid catalyst is further characterized by the T1/T2 ratio in 19F NMR relaxation measurements. The am-CN presents a higher surface affinity for the fluorinated substrate compared with other CN-based photocatalysts and thus shows the highest activity for this reaction [Figure 12D].
Beyond C-C coupling, the applications of supported metal NPs as photocatalysts have also been extended into N=N coupling reactions to produce azoxybenzenes and azobenzenes, which are important precursors for the dye, medical, and electronic industries. Zhu and coauthors reported the successful synthesis of azoxybenzenes and azobenzenes using supported Au and Cu NPs photocatalysts from nitrobenzene using isopropanol as the hydrogen source under visible light irradiation and basic conditions, as demonstrated in Equation (17)[189-191]:
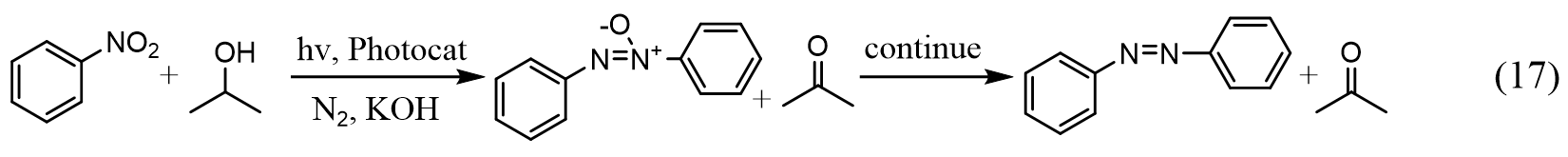
Here, the nitroaromatics undergo a gradual removal of oxygen by photogenerated protons and electrons via the formation of nitrosobenzene (NBS) and N-phenylhydroxylamine (NPH) radicals, which then generate azoxybenzene via dehydration coupling and eventually to azobenzene via removal of oxygen[192]. The selectivity of azoxybenzene or azobenzene can be tuned by controlling the chemical composition of the plasmonic metal NPs. While Au NPs supported on ZrO2 exhibit high selectivity towards azobenzene, the Ag-Cu alloy NPs supported on ZrO2 favor the selective formation of azoxybenzene. In addition, the selectivity can also be controlled by tuning the reaction temperature and employing Cu supported on graphene photocatalyst, though the activity decreases at low temperatures. These findings extend the potential application of the plasmonic metal NPs in photothermal catalysis for fine chemical synthesis; however, the high cost, low efficiency, and poor selectivity of such catalytic systems at RT limit their applications.
N=N coupling reaction can also be realized by using conventional semiconductor photocatalysts. Pal et al.[193] showed that the classical CdS quantum dots can be employed to reduce nitrobenzene to azoxybenzene with relatively high selectivity (68%) under visible light irradiation. It has been found that CdS with a small particle size (~2.8 nm) and the loading of Rh as cocatalyst are necessary to achieve a higher activity and selectivity towards azoxybenzene. Recently, our group showed that a series of azo- and azoxy-aromatic compounds can be selectively synthesized from their corresponding nitroaromatics via controlling the irradiation wavelength by simply using the pristine g-C3N4 under basic conditions with isopropanol as the hydrogen source[194]. Firstly, nitrobenzene can be converted into NBS and NPH. NBS and NPH are then further reduced to azoxy- and azo-aromatics. The azo-aromatics may be photoreduced into amines [Figure 13A]. While azobenzene is gradually formed in a two-step reduction of nitrobenzene under 410 nm irradiation with azoxybenzene as the intermediate [Figure 13B], azoxybenzene is the sole product throughout the 450 nm irradiation [Figure 13C]. Notably, high conversion of nitrobenzene (> 95%) and selectivity towards both desired products (> 94%) are reached for a series of nitroaromatics with different functional groups, which broaden its applications for the synthesis of complicated azo- and azoxy- compounds.

Figure 13. (A) Reaction path for the photoconversion of nitroaromatic compounds. (B,C) Light-tuned selective photosynthesis of azo- and azoxy-aromatics from nitrobenzene reduction using g-C3N4. (D) TPD spectra revealing the desorption of Hads from g-C3N4 and A-g-C3N4 surfaces. This figure is used with permission from a Creative Commons Attribution License[191]. (E) HRTEM image of the solid base Bi24O31Br10(OH)δ photocatalyst. (F) Basicity comparison of Bi24O31Br10(OH)δ, BiOBr, and g-C3N4 by CO2-TPD. (G) Photocatalytic selective nitrobenzene conversion under solar irradiation. This figure is used with permission from the American Chemistry Society[48]. TPD: Temperature-programmed desorption.
Moreover, the photocatalytic reductive coupling of nitroaromatics can be realized under solar irradiation at a large scale (80 L in volume) with high selectivity to azoxybenzene and a reasonable conversion. The origin of the activity is associated with the adsorption energy of surface adsorbed hydrogen atoms (Hads) that are generated by photo-oxidation of isopropanol, as confirmed by TPD of the pre-irradiated photocatalysts [Figure 13D]. While no desorption peak of H2 over active g-C3N4 indicates weakly bonded Hads on the catalyst surface, two obvious H2 desorption peaks observed over the inactive amorphous g-C3N4 (A-g-C3N4) suggest strong adsorption of the photogenerated Hads on A-g-C3N4. Therefore, weakly bonded Hads on g-C3N4 promotes the reduction of surface adsorbed nitrobenzene to form azo- or azoxy-benzene.
Although the g-C3N4 system shows great potential in photocatalytic reductive N=N coupling reactions, the low quantum efficiency (1.4% at 410 nm and 0.4% at 450 nm irradiation) and the need for a basic environment limit its application at a large scale. Since the photocatalytic hydrogen abstraction from alcohol is the initial step in reductive coupling, we expect that a photocatalyst coupled with engineered basic sites can improve the photocatalytic performance. Therefore, we developed basic-site-engineered bismuth oxybromide [Bi24O31Br10(OH)δ] nanosheets to boost nitrobenzene conversion under visible light irradiation and base-free conditions[48]. As presented in the HRTEM image of Figure 13E, the Bi24O31Br10(OH)δ nanosheets exhibit two types of lattice fringes (interplanar spacing of 0.278 and 0.299 nm) that match the (117) and (213) lattice planes of classical crystalline Bi24O31Br10, respectively. CO2-TPD reveals that the freshly prepared Bi24O31Br10(OH)δ nanosheets present a broad distribution of active sites with different basicities, whereas the typical BiOBr and g-C3N4 show almost no basicity [Figure 13F]. Indeed, the basic-site-engineered Bi24O31Br10(OH)δ nanosheets exhibit a robust hydrogen abstraction from isopropanol and fast hydrogen transfer from the catalyst to the adsorbed nitrobenzene according to in situ spectroscopy analysis, thus resulting in remarkable quantum efficiencies under visible light irradiation (42% at 410 nm and 32% at 450 nm). Nevertheless, the Bi24O31Br10(OH)δ nanosheets exhibit excellent performance in up-scaling and stability under solar irradiation. As depicted in Figure 13G, a complete conversion of nitrobenzene selectively to azoxybenzene and azobenzene (> 95%) in a sequential fashion is achieved under solar irradiation by employing the Bi24O31Br10(OH)δ photocatalyst, implying that the selective synthesis of two important products can simply be tuned via controlling irradiation time without the need of further separation process. In comparison, the g-C3N4 only converts ~20% of nitrobenzene under similar reaction conditions, which is less efficient than that of the Bi24O31Br10(OH)δ nanosheets.
Besides that, a very recent work shows that shape-engineered AgGaO2 delafossites with dominant (001) and (012) facets are efficient photocatalysts for efficient and selective photocatalytic redox coupling reactions under visible light [Figures 14A and B][195]. Efficient separation of the photogenerated charge carriers is observed over the two facets. The flat AgGaO2 (F-AGO) with electron-rich (001) facets shows an obvious enhancement over the stretched AgGaO2 (S-AGO) with hole-rich (012) facets in the reductive homocoupling of benzyl bromide and N-N coupling of nitrobenzene [Figure 14C]. In contrast, S-AGO performs better than F-AGO in the oxidative homocoupling of aniline and C-N coupling of amines [Figure 14D]. DFT calculation reveals that the difference in work functions of the (001) and (012) facets is the driving force for the tuned surface redox chemistry, which offers a noble metal-free strategy for the engineering of delafossite-based photocatalysts for synthetic chemistry.

Figure 14. (A,B) SEM of S-AGO and F-AGO. (C) Photocatalytic reductive coupling of benzyl bromide in isopropanol with 10 mM KOH under N2 conditions. (D) Photocatalytic oxidative coupling of 2,4,6-trimethylaniline in acetonitrile under aerated conditions. This figure is used with permission from the American Chemistry Society[195]. S-AGO: Stretched AgGaO2; F-AGO: flat AgGaO2.
Similar to the photocatalytic oxidative coupling reactions, the development of cross-coupling is far behind the homo-coupling investigations due to the lack of in-depth mechanistic understanding of reaction pathways. Thus, significant efforts should be paid to realize some representative cross-coupling reactions for applications.
CONCLUSION AND OUTLOOK
In this review, we summarize recent advances in photocatalytic synthesis from an application-orientated prospective to illustrate the development in the design of photocatalyst materials. Various reactions catalyzed by nanostructured photocatalysts are categorized into oxidative and reductive reactions, which are further subdivided into alcohol oxidation, epoxidation, oxidation of aromatics, oxidative coupling, hydrogenation reactions, and reductive coupling. The advantages and limitations of these nanostructured photocatalyst materials, including metal oxides, supported plasmonic metal nanostructures, organic polymers, anchored homogeneous catalysts, and dye-sensitized heterostructures, are presented. In addition, fundamental understandings of specific reaction mechanisms are demonstrated to guide the rational design of nanostructured photocatalysts.
Although many challenging reactions can be realized by employing various engineered heterogeneous photocatalyst materials, there are still critical challenges that need to be solved for scaling-up applications. First, a high selectivity, rather than a fast conversion rate, is crucial for all types of reactions from the industrial perspective, as the energy-consuming separation process may be eliminated. For photocatalytic oxidation processes, strong oxidants should be avoided to prevent over-oxidation. For the synthesis of complicated organic molecules, it is also desirable to have a control over the stereo-selectivity, which may be solved by anchoring the designed homogeneous redox catalyst with heterogeneous photocatalyst materials. The selectivity control calls for an in-depth understanding of specific reaction mechanisms to aid the design of engineered nanostructured photocatalyst materials. Besides, it is paramount to avoid using harsh reaction conditions and toxic compounds for reduction reactions (i.e., strong basic or acidic conditions, high temperature, strong reductants, and toxic solvents), as these diminish the advantages of the photocatalytic process. The mild reaction condition is also essential for recycling used photocatalysts and solvents. Nevertheless, the design and development of a continuous reactor (i.e., flow cell) customized for photocatalytic applications are urgently needed to evaluate photocatalyst materials and the scaling-up of photocatalytic processes.
DECLARATIONS
Authors’ contributionsPrepared the manuscript: Li Y, Su R
Corrected the manuscript: Zhang D, Qiao W, Xiang H, Besenbacher F, Li Y
Availability of data and materialsNot applicable.
Financial support and sponsorshipR.S. would like to thank the NSFC (projects number: 21972100) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, project number: NH10800120) for financial support. R. S. acknowledged Significant funding from Synfuels China Technology Co. Ltd.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Ertl G. Surface science and catalysis-studies on the mechanism of ammonia synthesis: The P. H. emmett award address. Catalysis Reviews 2006;21:201-23.
2. Yin H, Chen Z, Peng Y, et al. Dual active centers bridged by oxygen vacancies of ruthenium single-atom hybrids supported on molybdenum oxide for photocatalytic ammonia synthesis. Angew Chem Int Ed Engl 2022;61:e202114242.
4. Tuxen A, Kibsgaard J, Gøbel H, et al. Size threshold in the dibenzothiophene adsorption on MoS2 nanoclusters. ACS Nano 2010;4:4677-82.
5. Su R, Lü Z, Chen K, et al. Novel in situ method (vacuum assisted electroless plating) modified porous cathode for solid oxide fuel cells. Electrochemistry Communications 2008;10:844-7.
6. Fujishima A, Honda K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972;238:37-8.
7. Sunada K, Watanabe T, Hashimoto K. Studies on photokilling of bacteria on TiO2 thin film. Journal of Photochemistry and Photobiology A: Chemistry 2003;156:227-33.
8. Su R, Dimitratos N, Liu J, et al. Mechanistic insight into the interaction between a titanium dioxide photocatalyst and Pd cocatalyst for improved photocatalytic performance. ACS Catal 2016;6:4239-47.
9. Su R, Tiruvalam R, He Q, et al. Promotion of phenol photodecomposition over TiO2 using Au, Pd, and Au-Pd nanoparticles. ACS Nano 2012;6:6284-92.
10. Wang X, Sø L, Su R, et al. The influence of crystallite size and crystallinity of anatase nanoparticles on the photo-degradation of phenol. Journal of Catalysis 2014;310:100-8.
11. Goto Y, Hisatomi T, Wang Q, et al. A particulate photocatalyst water-splitting panel for large-scale solar hydrogen generation. Joule 2018;2:509-20.
13. Liu N, Schneider C, Freitag D, et al. Black TiO2 nanotubes: cocatalyst-free open-circuit hydrogen generation. Nano Lett 2014;14:3309-13.
14. Cao J, Ren L, Li N, Hu C, Cao M. Mesoporous Ta(3)N(5) microspheres prepared from a high-surface-area, microporous, amorphous precursor and their visible-light-driven photocatalytic activity. Chemistry 2013;19:12619-23.
15. Burton LA, Colombara D, Abellon RD, et al. Synthesis, characterization, and electronic structure of single-crystal SnS, Sn2S3, and SnS2. Chem Mater 2013;25:4908-16.
16. Wang Y, Zhang Z, Zhu Y, et al. Nanostructured VO2 photocatalysts for hydrogen production. ACS Nano 2008;2:1492-6.
17. Bao N, Shen L, Takata T, Domen K. Self-templated synthesis of nanoporous CdS nanostructures for highly efficient photocatalytic hydrogen production under visible light. Chem Mater 2008;20:110-7.
18. Das R, Sarkar S, Kumar R, et al. Noble-metal-free heterojunction photocatalyst for selective CO2 reduction to methane upon induced strain relaxation. ACS Catal 2022;12:687-97.
19. Liu Q, Wang S, Mo W, et al. Emerging stacked photocatalyst design enables spatially separated Ni(OH)2 redox cocatalysts for overall CO2 reduction and H2O oxidation. Small 2022;18:e2104681.
20. Chen Y, Zhang Y, Fan G, et al. Cooperative catalysis coupling photo-/photothermal effect to drive Sabatier reaction with unprecedented conversion and selectivity. Joule 2021;5:3235-51.
21. Ghasimi S, Prescher S, Wang ZJ, Landfester K, Yuan J, Zhang KA. Heterophase photocatalysts from water-soluble conjugated polyelectrolytes: an example of self-initiation under visible light. Angew Chem Int Ed Engl 2015;54:14549-53.
22. Zhang H, Liu G, Shi L, Liu H, Wang T, Ye J. Engineering coordination polymers for photocatalysis. Nano Energy 2016;22:149-68.
23. Zhang G, Liu G, Wang L, Irvine JT. Inorganic perovskite photocatalysts for solar energy utilization. Chem Soc Rev 2016;45:5951-84.
24. Dong B, Cui J, Liu T, et al. Development of novel perovskite-like oxide photocatalyst LiCuTa3O9 with dual functions of water reduction and oxidation under visible light irradiation. Adv Energy Mater 2018;8:1801660.
25. Wang J, Wang C, Lin W. Metal-organic frameworks for light harvesting and photocatalysis. ACS Catal 2012;2:2630-40.
26. Wang S, Wang X. Multifunctional metal-organic frameworks for photocatalysis. Small 2015;11:3097-112.
27. Fu S, Yao S, Guo S, et al. Feeding carbonylation with CO2 via the synergy of single-site/nanocluster catalysts in a photosensitizing MOF. J Am Chem Soc 2021;143:20792-801.
28. Cheng X, Dao X, Wang S, Zhao J, Sun W. Enhanced photocatalytic CO2 reduction activity over NH2-MIL-125(Ti) by facet regulation. ACS Catal 2021;11:650-8.
29. Lu H, Hao Q, Chen T, et al. A high-performance Bi2O3/Bi2SiO5 p-n heterojunction photocatalyst induced by phase transition of Bi2O3. Applied Catalysis B: Environmental 2018;237:59-67.
30. Low J, Yu J, Jaroniec M, Wageh S, Al-Ghamdi AA. Heterojunction photocatalysts. Adv Mater 2017;29:1601694.
31. Yang Y, Sun C, Wang L, et al. Constructing a metallic/semiconducting TaB2/Ta2O5 core/shell heterostructure for photocatalytic hydrogen evolution. Adv Energy Mater 2014;4:1400057.
32. Hu J, Chen D, Mo Z, et al. Z-scheme 2D/2D heterojunction of black phosphorus/monolayer Bi2WO6 nanosheets with enhanced photocatalytic activities. Angew Chem Int Ed Engl 2019;58:2073-7.
33. Yang J, Wang D, Han H, Li C. Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc Chem Res 2013;46:1900-9.
34. Maeda K, Abe R, Domen K. Role and function of ruthenium species as promoters with TaON-based photocatalysts for oxygen evolution in two-step water splitting under Visible Light. J Phys Chem C 2011;115:3057-64.
35. Vercammen J, Bocus M, Neale S, et al. Shape-selective C-H activation of aromatics to biarylic compounds using molecular palladium in zeolites. Nat Catal 2020;3:1002-9.
36. Wang Y, Lv H, Grape ES, et al. A tunable multivariate metal-organic framework as a platform for designing photocatalysts. J Am Chem Soc 2021;143:6333-8.
37. Rosso C, Filippini G, Prato M. Use of nitrogen-doped carbon nanodots for the photocatalytic fluoroalkylation of organic compounds. Chemistry ;2019:16032-6.
38. Zhao X, Deng C, Meng D, et al. Nickel-coordinated carbon nitride as a metallaphotoredox platform for the cross-coupling of aryl halides with alcohols. ACS Catal 2020;10:15178-85.
39. Huang Y, Liu C, Li M, et al. Photoimmobilized Ni clusters boost photodehydrogenative coupling of amines to imines via enhanced hydrogen evolution kinetics. ACS Catal 2020;10:3904-10.
40. Li S, Kim S, Davis AH, et al. Photocatalytic chemoselective C-C bond cleavage at room temperature in dye-sensitized photoelectrochemical cells. ACS Catal 2021;11:3771-81.
41. Mazzanti S, Kurpil B, Pieber B, Antonietti M, Savateev A. Dichloromethylation of enones by carbon nitride photocatalysis. Nat Commun 2020;11:1387.
42. Khamrai J, Ghosh I, Savateev A, Antonietti M, König B. Photo-Ni-dual-catalytic C(sp2)-C(sp3) cross-coupling reactions with mesoporous graphitic carbon nitride as a heterogeneous organic semiconductor photocatalyst. ACS Catal 2020;10:3526-32.
43. Ithisuphalap K, Zhang H, Guo L, Yang Q, Yang H, Wu G. Photocatalysis and photoelectrocatalysis methods of nitrogen reduction for sustainable ammonia synthesis. Small Methods 2019;3:1800352.
44. Zhao Y, Zhao Y, Waterhouse GIN, et al. Layered-double-hydroxide nanosheets as efficient visible-light-driven photocatalysts for dinitrogen fixation. Adv Mater 2017;29:1703828.
45. Chen L, Tang J, Song L, et al. Heterogeneous photocatalysis for selective oxidation of alcohols and hydrocarbons. Applied Catalysis B: Environmental 2019;242:379-88.
46. Parrino F, Bellardita M, García-lópez EI, Marcì G, Loddo V, Palmisano L. Heterogeneous photocatalysis for selective formation of high-value-added molecules: some chemical and engineering aspects. ACS Catal 2018;8:11191-225.
47. Huang Y, Liu Z, Gao G, et al. Stable copper nanoparticle photocatalysts for selective epoxidation of alkenes with visible light. ACS Catal 2017;7:4975-85.
48. Dai Y, Li C, Shen Y, et al. Efficient solar-driven hydrogen transfer by bismuth-based photocatalyst with engineered basic sites. J Am Chem Soc 2018;140:16711-9.
49. Ma D, Liu A, Li S, Lu C, Chen C. TiO2 photocatalysis for C-C bond formation. Catal Sci Technol 2018;8:2030-45.
50. Fagnoni M, Dondi D, Ravelli D, Albini A. Photocatalysis for the formation of the C-C bond. Chem Rev 2007;107:2725-56.
51. Corrigan N, Shanmugam S, Xu J, Boyer C. Photocatalysis in organic and polymer synthesis. Chem Soc Rev 2016;45:6165-212.
52. Chen TQ, MacMillan DWC. A metallaphotoredox strategy for the cross-Electrophile coupling of α-Chloro carbonyls with aryl halides. Angew Chem Int Ed Engl 2019;58:14584-8.
53. Vasilopoulos A, Krska SW, Stahl SS. C(sp3)-H methylation enabled by peroxide photosensitization and Ni-mediated radical coupling. Science 2021;372:398-403.
54. Liu Z, Nan X, Lei T, et al. Photo-induced reductive cross-coupling of aldehydes, ketones and imines with electron-deficient arenes to construct aryl substituted alcohols and amines. Chinese Journal of Catalysis 2018;39:487-94.
55. Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DW. Switching on elusive organometallic mechanisms with photoredox catalysis. Nature 2015;524:330-4.
56. Nakajima M, Fava E, Loescher S, Jiang Z, Rueping M. Photoredox-Catalyzed Reductive Coupling of Aldehydes, Ketones, and Imines with Visible Light. Angew Chem Int Ed Engl 2015;54:8828-32.
57. Zhao G, Yang C, Guo L, Sun H, Lin R, Xia W. Reactivity insight into reductive coupling and aldol cyclization of chalcones by visible light photocatalysis. J Org Chem 2012;77:6302-6.
58. Marzo L, Pagire SK, Reiser O, König B. Visible-light photocatalysis: does it make a difference in organic synthesis? Angew Chem Int Ed Engl 2018;57:10034-72.
59. Dong Z, MacMillan DWC. Metallaphotoredox-enabled deoxygenative arylation of alcohols. Nature 2021;598:451-6.
60. Torres GM, Liu Y, Arndtsen BA. A dual light-driven palladium catalyst: Breaking the barriers in carbonylation reactions. Science 2020;368:318-23.
61. Xu B, Troian-Gautier L, Dykstra R, Martin RT, Gutierrez O, Tambar UK. Photocatalyzed diastereoselective isomerization of cinnamyl chlorides to cyclopropanes. J Am Chem Soc 2020;142:6206-15.
62. Yang Q, Wang YH, Qiao Y, et al. Photocatalytic C-H activation and the subtle role of chlorine radical complexation in reactivity. Science 2021;372:847-52.
63. Constantin T, Zanini M, Regni A, Sheikh NS, Juliá F, Leonori D. Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 2020;367:1021-6.
64. Lee GS, Kim D, Hong SH. Pd-catalyzed formal Mizoroki-Heck coupling of unactivated alkyl chlorides. Nat Commun 2021;12:991.
65. Chen C, Peters JC, Fu GC. Photoinduced copper-catalysed asymmetric amidation via ligand cooperativity. Nature 2021;596:250-6.
66. Suzuki K, Mizuno N, Yamaguchi K. Polyoxometalate photocatalysis for liquid-phase selective organic functional group transformations. ACS Catal 2018;8:10809-25.
67. Kobielusz M, Mikrut P, Macyk W. Materials for sustainable energy. Available from: https://www.elsevier.com/books/materials-for-sustainable-energy/van-eldik/978-0-12-815077-1 [Last accessed on 21 Apr 2022].
68. Miyabe H, Kohtani S. Photocatalytic single electron transfer reactions on TiO2 semiconductor. Sci China Chem 2019;62:1439-49.
69. Bloh JZ, Marschall R. Heterogeneous photoredox catalysis: reactions, materials, and reaction engineering. Eur J Org Chem 2017;2017:2085-94.
70. Chen Y, Lu L, Yu D, Zhu C, Xiao W. Visible light-driven organic photochemical synthesis in China. Sci China Chem 2019;62:24-57.
71. Colmenares JC, Luque R. Heterogeneous photocatalytic nanomaterials: prospects and challenges in selective transformations of biomass-derived compounds. Chem Soc Rev 2014;43:765-78.
72. Fang Y, Zheng Y, Fang T, et al. Photocatalysis: an overview of recent developments and technological advancements. Sci China Chem 2020;63:149-81.
73. Cheng H, Xu W. Recent advances in modified TiO2 for photo-induced organic synthesis. Org Biomol Chem 2019;17:9977-89.
74. Gisbertz S, Pieber B. Heterogeneous photocatalysis in organic synthesis. ChemPhotoChem 2020;4:456-75.
75. Bamwenda GR, Tsubota S, Nakamura T, Haruta M. Photoassisted hydrogen production from a water-ethanol solution: a comparison of activities of Au-TiO2 and Pt-TiO2. Journal of Photochemistry and Photobiology A: Chemistry 1995;89:177-89.
76. Bahruji H, Bowker M, Davies PR, Pedrono F. New insights into the mechanism of photocatalytic reforming on Pd/TiO2. Applied Catalysis B: Environmental 2011;107:205-9.
77. Wang X, Maeda K, Thomas A, et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat Mater 2009;8:76-80.
78. Wu B, Liu D, Mubeen S, Chuong TT, Moskovits M, Stucky GD. Anisotropic growth of TiO2 onto gold nanorods for plasmon-enhanced hydrogen production from water reduction. J Am Chem Soc 2016;138:1114-7.
79. Samanta S, Martha S, Parida K. Facile synthesis of Au/g-C3N4 nanocomposites: an inorganic/organic hybrid plasmonic photocatalyst with enhanced hydrogen gas evolution under visible-light irradiation. ChemCatChem 2014; doi: 10.1002/cctc.201300949.
80. Ruberu TPA, Nelson NC, Slowing II, Vela J. Selective alcohol dehydrogenation and hydrogenolysis with semiconductor-metal photocatalysts: toward solar-to-chemical energy conversion of biomass-relevant substrates. J Phys Chem Lett 2012;3:2798-802.
81. Lu H, Zhao J, Li L, et al. Selective oxidation of sacrificial ethanol over TiO2-based photocatalysts during water splitting. Energy Environ Sci 2011;4:3384.
82. Zhang H, Wu Y, Li L, Zhu Z. Photocatalytic direct conversion of ethanol to 1,1- diethoxyethane over noble-metal-loaded TiO2 nanotubes and nanorods. ChemSusChem 2015;8:1226-31.
83. Pillai UR, Sahle–demessie E. Selective oxidation of alcohols in gas phase using light-activated titanium dioxide. Journal of Catalysis 2002;211:434-44.
84. Park JY, Baker LR, Somorjai GA. Role of hot electrons and metal-oxide interfaces in surface chemistry and catalytic reactions. Chem Rev 2015;115:2781-817.
85. Li H, Qin F, Yang Z, Cui X, Wang J, Zhang L. New reaction pathway induced by plasmon for selective benzyl alcohol oxidation on BiOCl possessing oxygen vacancies. J Am Chem Soc 2017;139:3513-21.
86. Nakato Y, Ueda K, Yano H, Tsubomura H. Effect of microscopic discontinuity of metal overlayers on the photovoltages in metal-coated semiconductor-liquid junction photoelectrochemical cells for efficient solar energy conversion. J Phys Chem 1988;92:2316-24.
87. Sakamoto H, Ohara T, Yasumoto N, et al. Hot-Electron-Induced Highly Efficient O2 Activation by Pt Nanoparticles Supported on Ta2O5 Driven by Visible Light. J Am Chem Soc 2015;137:9324-32.
88. Sugano Y, Shiraishi Y, Tsukamoto D, Ichikawa S, Tanaka S, Hirai T. Supported Au-Cu bimetallic alloy nanoparticles: an aerobic oxidation catalyst with regenerable activity by visible-light irradiation. Angew Chem Int Ed Engl 2013;52:5295-9.
89. Landry MJ, Gellé A, Meng BY, Barrett CJ, Moores A. Surface-plasmon-mediated hydrogenation of carbonyls catalyzed by silver nanocubes under visible light. ACS Catal 2017;7:6128-33.
90. Han C, Yang X, Gao G, et al. Selective oxidation of methanol to methyl formate on catalysts of Au-Ag alloy nanoparticles supported on titania under UV irradiation. Green Chem 2014;16:3603-15.
91. Xiao Q, Liu Z, Bo A, et al. Catalytic transformation of aliphatic alcohols to corresponding esters in O2 under neutral conditions using visible-light irradiation. J Am Chem Soc 2015;137:1956-66.
92. Su R, Tiruvalam R, Logsdail AJ, et al. Designer titania-supported Au-Pd nanoparticles for efficient photocatalytic hydrogen production. ACS Nano 2014;8:3490-7.
93. Jiang X, Fu X, Zhang L, Meng S, Chen S. Photocatalytic reforming of glycerol for H2 evolution on Pt/TiO2: fundamental understanding the effect of co-catalyst Pt and the Pt deposition route. J Mater Chem A 2015;3:2271-82.
94. Sanwald KE, Berto TF, Eisenreich W, Gutiérrez OY, Lercher JA. Catalytic routes and oxidation mechanisms in photoreforming of polyols. Journal of Catalysis 2016;344:806-16.
95. Berto TF, Sanwald KE, Eisenreich W, Gutiérrez OY, Lercher JA. Photoreforming of ethylene glycol over Rh/TiO2 and Rh/GaN:ZnO. Journal of Catalysis 2016;338:68-81.
96. Jin X, Li C, Xu C, et al. Photocatalytic C C bond cleavage in ethylene glycol on TiO2: a molecular level picture and the effect of metal nanoparticles. Journal of Catalysis 2017;354:37-45.
97. Chong R, Li J, Zhou X, et al. Selective photocatalytic conversion of glycerol to hydroxyacetaldehyde in aqueous solution on facet tuned TiO2-based catalysts. Chem Commun (Camb) 2014;50:165-7.
98. Augugliaro V, El Nazer HH, Loddo V, et al. Partial photocatalytic oxidation of glycerol in TiO2 water suspensions. Catalysis Today 2010;151:21-8.
99. Li C, Wang X, Cheruvathur A, et al. In-situ probing photocatalytic C-C bond cleavage in ethylene glycol under ambient conditions and the effect of metal cocatalyst. Journal of Catalysis 2018;365:313-9.
100. Zhang Y, Zhang N, Tang Z, Xu Y. Identification of Bi2WO6 as a highly selective visible-light photocatalyst toward oxidation of glycerol to dihydroxyacetone in water. Chem Sci 2013;4:1820.
101. Sajkowski DJ, Boudart M. Structure sensitivity of the catalytic oxidation of ethene by silver. Catalysis Reviews 1987;29:325-60.
102. Nijhuis TA, Makkee M, Moulijn JA, Weckhuysen BM. The Production of propene oxide: catalytic processes and recent developments. Ind Eng Chem Res 2006;45:3447-59.
103. Torres D, Lopez N, Illas F, Lambert RM. Why copper is intrinsically more selective than silver in alkene epoxidation: ethylene oxidation on Cu(111) versus Ag(111). J Am Chem Soc 2005;127:10774-5.
104. Christopher P, Xin H, Linic S. Visible-light-enhanced catalytic oxidation reactions on plasmonic silver nanostructures. Nat Chem 2011;3:467-72.
105. Christopher P, Xin H, Marimuthu A, Linic S. Singular characteristics and unique chemical bond activation mechanisms of photocatalytic reactions on plasmonic nanostructures. Nat Mater 2012;11:1044-50.
106. Marimuthu A, Zhang J, Linic S. Tuning selectivity in propylene epoxidation by plasmon mediated photo-switching of Cu oxidation state. Science 2013;339:1590-3.
107. Zhang X, Kumari G, Heo J, Jain PK. In situ formation of catalytically active graphene in ethylene photo-epoxidation. Nat Commun 2018;9:3056.
108. Jin JK, Wu K, Liu XY, et al. Building a Pyrazole-benzothiadiazole-pyrazole photosensitizer into metal-organic frameworks for photocatalytic aerobic oxidation. J Am Chem Soc 2021;143:21340-9.
109. Dai Y, Ren P, Li Y, et al. Solid base Bi24O31Br10(OH)δ with Active lattice oxygen for the efficient photo-oxidation of primary alcohols to aldehydes. Angew Chem Int Ed Engl 2019;58:6265-70.
110. Chen X, Zhang J, Fu X, Antonietti M, Wang X. Fe-g-C3N4-catalyzed oxidation of benzene to phenol using hydrogen peroxide and visible light. J Am Chem Soc 2009;131:11658-9.
111. Yoshida H, Yuzawa H, Aoki M, Otake K, Itoh H, Hattori T. Photocatalytic hydroxylation of aromatic ring by using water as an oxidant. Chem Commun (Camb) 2008:4634-6.
112. Yuzawa H, Aoki M, Otake K, Hattori T, Itoh H, Yoshida H. Reaction mechanism of aromatic ring hydroxylation by water over platinum-loaded titanium oxide photocatalyst. J Phys Chem C 2012;116:25376-87.
113. Zhang G, Yi J, Shim J, Lee J, Choi W. Photocatalytic hydroxylation of benzene to phenol over titanium oxide entrapped into hydrophobically modified siliceous foam. Applied Catalysis B: Environmental 2011;102:132-9.
114. Su R, Kesavan L, Jensen MM, et al. Selective photocatalytic oxidation of benzene for the synthesis of phenol using engineered Au-Pd alloy nanoparticles supported on titanium dioxide. Chem Commun (Camb) 2014;50:12612-4.
115. Hosseini SM, Ghiaci M, Kulinich SA, et al. Au-Pd@g-C3N4 as an efficient photocatalyst for visible-light oxidation of benzene to phenol: experimental and mechanistic study. J Phys Chem C 2018;122:27477-85.
116. He J, Zhang M, Primo A, García H, Li Z. Selective photocatalytic benzene hydroxylation to phenol using surface-modified Cu2O supported on graphene. J Mater Chem A 2018;6:19782-7.
117. Ide Y, Matsuoka M, Ogawa M. Efficient visible-light-induced photocatalytic activity on gold-nanoparticle-supported layered titanate. J Am Chem Soc 2010;132:16762-4.
118. Jaynes BS, Hill CL. Radical carbonylation of alkanes via polyoxotungstate photocatalysis. J Am Chem Soc 1995;117:4704-5.
119. Suzuki K, Tang F, Kikukawa Y, Yamaguchi K, Mizuno N. Visible-light-induced photoredox catalysis with a tetracerium-containing silicotungstate. Angew Chem Int Ed Engl 2014;53:5356-60.
120. Yamamoto A, Ohara T, Yoshida H. Visible-light-induced photocatalytic benzene/cyclohexane cross-coupling utilizing a ligand-to-metal charge transfer benzene complex adsorbed on titanium oxides. Catal Sci Technol 2018;8:2046-50.
121. Cao X, Chen Z, Lin R, et al. A photochromic composite with enhanced carrier separation for the photocatalytic activation of benzylic C-H bonds in toluene. Nat Catal 2018;1:704-10.
122. Wu X, Fan X, Xie S, et al. Solar energy-driven lignin-first approach to full utilization of lignocellulosic biomass under mild conditions. Nat Catal 2018;1:772-80.
123. Eisenhofer A, Hioe J, Gschwind RM, König B. Photocatalytic phenol-arene C-C and C-O cross-dehydrogenative coupling: photocatalytic phenol-arene C-C and C-O cross-dehydrogenative coupling. Eur J Org Chem 2017;2017:2194-204.
124. Zhao G, Yang C, Guo L, Sun H, Chen C, Xia W. Visible light-induced oxidative coupling reaction: easy access to Mannich-type products. Chem Commun (Camb) 2012;48:2337-9.
125. Tan Y, Lin S, Liu C, et al. Boosting photocatalytic cross-dehydrogenative coupling reaction by incorporating [RuII(bpy)3] into a radical metal-organic framework. Applied Catalysis B: Environmental 2018;227:425-32.
126. Zhang N, Li X, Ye H, et al. Oxide defect engineering enables to couple solar energy into oxygen activation. J Am Chem Soc 2016;138:8928-35.
127. Raza F, Park JH, Lee H, Kim H, Jeon S, Kim J. Visible-light-driven oxidative coupling reactions of amines by photoactive WS2 nanosheets. ACS Catal 2016;6:2754-9.
128. Xu Y, Chen Y, Fu W. Visible-light driven oxidative coupling of amines to imines with high selectivity in air over core-shell structured CdS@C3N4. Applied Catalysis B: Environmental 2018;236:176-83.
129. Yu J, Liu Q, Qiao W, et al. Catalytic role of metal nanoparticles in selectivity control over photodehydrogenative coupling of primary amines to imines and secondary amines. ACS Catal 2021;11:6656-61.
130. Xie S, Shen Z, Deng J, et al. Visible light-driven C-H activation and C-C coupling of methanol into ethylene glycol. Nat Commun 2018;9:1181.
131. Xiao Q, Sarina S, Bo A, et al. Visible light-driven cross-coupling reactions at lower temperatures using a photocatalyst of palladium and gold alloy nanoparticles. ACS Catal 2014;4:1725-34.
132. Ghasimi S, Bretschneider SA, Huang W, Landfester K, Zhang KAI. A Conjugated microporous polymer for palladium-free, visible light-promoted photocatalytic stille-type coupling reactions. Adv Sci (Weinh) 2017;4:1700101.
133. Shiraishi Y, Fujiwara K, Sugano Y, Ichikawa S, Hirai T. N-Monoalkylation of amines with alcohols by tandem photocatalytic and catalytic reactions on TiO2 loaded with Pd nanoparticles. ACS Catal 2013;3:312-20.
134. Wang LM, Morioka Y, Jenkinson K, Wheatley AEH, Saito S, Naka H. N-Alkylation of functionalized amines with alcohols using a copper-gold mixed photocatalytic system. Sci Rep 2018;8:6931.
135. Lv D, Li Y, Qiao W, et al. Metal cocatalyst mediated photocatalytic dehydrogenative-condensation and direct condensation cross-coupling of aniline and alcohol. Applied Catalysis B: Environmental 2022;309:121264.
136. Alkhatib II, Garlisi C, Pagliaro M, Al-ali K, Palmisano G. Metal-organic frameworks for photocatalytic CO2 reduction under visible radiation: a review of strategies and applications. Catalysis Today 2020;340:209-24.
137. Yu S, Wilson AJ, Heo J, Jain PK. Plasmonic control of multi-electron transfer and C-C coupling in visible-light-driven CO2 reduction on Au nanoparticles. Nano Lett 2018;18:2189-94.
138. Iwase A, Yoshino S, Takayama T, Ng YH, Amal R, Kudo A. Water splitting and CO2 reduction under visible light irradiation using Z-scheme systems consisting of metal sulfides, CoOx-loaded BiVO4, and a reduced graphene oxide electron mediator. J Am Chem Soc 2016;138:10260-4.
139. Wang S, Hou Y, Wang X. Development of a stable MnCo2O4 cocatalyst for photocatalytic CO2 reduction with visible light. ACS Appl Mater Interfaces 2015;7:4327-35.
140. Ye M, Wang X, Liu E, Ye J, Wang D. Boosting the photocatalytic activity of P25 for carbon dioxide reduction by using a surface-alkalinized titanium carbide mxene as cocatalyst. ChemSusChem 2018;11:1606-11.
141. Iizuka K, Wato T, Miseki Y, Saito K, Kudo A. Photocatalytic reduction of carbon dioxide over Ag cocatalyst-loaded ALa4Ti4O15 (A = Ca, Sr, and Ba) using water as a reducing reagent. J Am Chem Soc 2011;133:20863-8.
142. Chang X, Wang T, Gong J. CO2 photo-reduction: insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ Sci 2016;9:2177-96.
143. Sastre F, Corma A, García H. Visible-light photocatalytic conversion of carbon monoxide to methane by nickel(II) oxide. Angew Chem Int Ed Engl 2013;52:12983-7.
144. Sastre F, Puga AV, Liu L, Corma A, García H. Complete photocatalytic reduction of CO2 to methane by H2 under solar light irradiation. J Am Chem Soc 2014;136:6798-801.
145. Guo X, Jiao Z, Jin G, Guo X. Photocatalytic Fischer-Tropsch synthesis on graphene-supported worm-like ruthenium nanostructures. ACS Catal 2015;5:3836-40.
146. Chen G, Gao R, Zhao Y, et al. Alumina-supported CoFe Alloy catalysts derived from layered-double-hydroxide nanosheets for efficient photothermal CO2 hydrogenation to hydrocarbons. Adv Mater 2018;30:1704663.
147. Zhao Y, Li Z, Li M, et al. Reductive transformation of layered-double-hydroxide nanosheets to fe-based heterostructures for efficient visible-light photocatalytic hydrogenation of CO. Adv Mater 2018:e1803127.
148. Li Z, Zhang X, Liu J, et al. Titania-supported Ni2P/Ni catalysts for selective solar-driven CO hydrogenation. Adv Mater 2021;33:e2103248.
149. Li Z, Liu J, Shi R, Waterhouse GIN, Wen X, Zhang T. Fe-based catalysts for the direct photohydrogenation of CO2 to value-added hydrocarbons. Adv Energy Mater 2021;11:2002783.
150. Ye JH, Ju T, Huang H, Liao LL, Yu DG. Radical Carboxylative Cyclizations and Carboxylations with CO2. Acc Chem Res 2021;54:2518-31.
151. Yang T, Yu Q, Wang H. Photocatalytic reduction of CO2 to CH3OH coupling with the oxidation of amine to imine. Catal Lett 2018;148:2382-90.
152. Chen Y, Wang M, Ma Y, Li Y, Cai J, Li Z. Coupling photocatalytic CO2 reduction with benzyl alcohol oxidation to produce benzyl acetate over Cu2O/Cu. Catal Sci Technol 2018;8:2218-23.
153. Campos-Martin JM, Blanco-Brieva G, Fierro JL. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew Chem Int Ed Engl 2006;45:6962-84.
154. Teranishi M, Naya S, Tada H. In situ liquid phase synthesis of hydrogen peroxide from molecular oxygen using gold nanoparticle-loaded titanium(IV) dioxide photocatalyst. J Am Chem Soc 2010;132:7850-1.
155. Xiong X, Zhang X, Liu S, Zhao J, Xu Y. Sustained production of H2O2 in alkaline water solution using borate and phosphate-modified Au/TiO2 photocatalysts. Photochem Photobiol Sci 2018;17:1018-22.
156. Moon G, Kim W, Bokare AD, Sung N, Choi W. Solar production of H2O2 on reduced graphene oxide-TiO2 hybrid photocatalysts consisting of earth-abundant elements only. Energy Environ Sci 2014;7:4023-8.
157. Shiraishi Y, Kanazawa S, Sugano Y, et al. Highly selective production of hydrogen peroxide on graphitic carbon nitride (g-C3N4) photocatalyst activated by visible light. ACS Catal 2014;4:774-80.
158. He B, Feng M, Chen X, Sun J. Multidimensional (0D-3D) Functional nanocarbon: promising material to strengthen the photocatalytic activity of graphitic carbon nitride. Green Energy Environ 2021;6:823-45.
159. Li S, Dong G, Hailili R, et al. Effective photocatalytic H2O2 production under visible light irradiation at g-C3N4 modulated by carbon vacancies. Applied Catalysis B: Environmental 2016;190:26-35.
160. Kim H, Choi Y, Hu S, Choi W, Kim J. Photocatalytic hydrogen peroxide production by anthraquinone-augmented polymeric carbon nitride. Applied Catalysis B: Environmental 2018;229:121-9.
161. Teng Z, Zhang Q, Yang H, et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat Catal 2021;4:374-84.
162. Joyce-pruden C, Pross JK, Li Y. Photoinduced reduction of aldehydes on titanium dioxide. J Org Chem 1992;57:5087-91.
163. Yanagida S, Yoshiya M, Shiragami T, Pac C, Mori H, Fujita H. Semiconductor photocatalysis. I. Quantitative photoreduction of aliphatic ketones to alcohols using defect-free zinc sulfide quantum crystallites. J Phys Chem 1990;94:3104-11.
164. Hao D, Liu Y, Gao S, et al. Emerging artificial nitrogen cycle processes through novel electrochemical and photochemical synthesis. Materials Today 2021;46:212-33.
165. Zhang H, Zhao M, Zhao T, Li L, Zhu Z. Hydrogenative cyclization of levulinic acid into γ-valerolactone by photocatalytic intermolecular hydrogen transfer. Green Chem 2016;18:2296-301.
166. Kohtani S, Nishioka S, Yoshioka E, Miyabe H. Dye-sensitized photo-hydrogenation of aromatic ketones on titanium dioxide under visible light irradiation. Catalysis Communications 2014;43:61-5.
167. Jiao Z, Guo X, Zhai Z, Jin G, Wang X, Guo X. The enhanced catalytic performance of Pd/SiC for the hydrogenation of furan derivatives at ambient temperature under visible light irradiation. Catal Sci Technol 2014;4:2494-8.
168. Hao CH, Guo XN, Pan YT, et al. Visible-light-driven selective photocatalytic hydrogenation of cinnamaldehyde over Au/SiC catalysts. J Am Chem Soc 2016;138:9361-4.
169. Shiraishi Y, Togawa Y, Tsukamoto D, Tanaka S, Hirai T. Highly efficient and selective hydrogenation of nitroaromatics on photoactivated rutile titanium dioxide. ACS Catal 2012;2:2475-81.
170. Toyao T, Saito M, Horiuchi Y, et al. Efficient hydrogen production and photocatalytic reduction of nitrobenzene over a visible-light-responsive metal–organic framework photocatalyst. Catal Sci Technol 2013;3:2092.
171. Gao WZ, Xu Y, Chen Y, Fu WF. Highly efficient and selective photocatalytic reduction of nitroarenes using the Ni2P/CdS catalyst under visible-light irradiation. Chem Commun (Camb) 2015;51:13217-20.
172. Xiao G, Li P, Zhao Y, Xu S, Su H. Visible-light-driven chemoselective hydrogenation of nitroarenes to anilines in water through graphitic carbon nitride metal-free photocatalysis. Chem Asian J ;2018:1950-5.
173. Huang Y, Liu Z, Gao G, et al. Visible light-driven selective hydrogenation of unsaturated aromatics in an aqueous solution by direct photocatalysis of Au nanoparticles. Catal Sci Technol 2018;8:726-34.
174. Buckler SA, Doll L, Lind FK, Epstein M. Phosphine as a reducing agent. J Org Chem 1962;27:794-8.
175. Galbraith HW, Degering EF, Hitch EF. The Alkaline reduction of aromatic nitro compounds with glucose. J Am Chem Soc 1951;73:1323-4.
176. Mckillop A, Raphael RA, Taylor EC. Thallium in organic synthesis. XI. Preparation of azoxy compounds. J Org Chem 1970;35:1670-2.
177. Osuka A, Shimizu H, Suzuki H. Reduction of aromatic and aliphatic Nitro compounds by sodium hydrogen telluride. Chem Lett 1983;12:1373-4.
178. Fortman GC, Nolan SP. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: a perfect union. Chem Soc Rev 2011;40:5151-69.
179. Zhang G, Scott BL, Hanson SK. Mild and homogeneous cobalt-catalyzed hydrogenation of C=C, C=O, and C=N bonds. Angew Chem Int Ed Engl 2012;51:12102-6.
180. Zhang X, Smith RT, Le C, et al. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 2020;580:220-6.
181. Laudadio G, Deng Y, van der Wal K, et al. C(sp3)-H functionalizations of light hydrocarbons using decatungstate photocatalysis in flow. Science 2020;369:92-6.
182. Dong XY, Cheng JT, Zhang YF, et al. Copper-catalyzed asymmetric radical 1,2-carboalkynylation of alkenes with alkyl halides and terminal alkynes. J Am Chem Soc 2020;142:9501-9.
183. Li Z, Jin J, Huang S. Recent advances in transition metal-catalyzed cross-coupling reactions directly promoted by visible light. Chin J Org Chem 2020;40:563.
184. Zhang Z, Xu Y, Zhang Q, et al. Semi-heterogeneous photo-Cu-dual-catalytic cross-coupling reactions using polymeric carbon nitrides. Science Bulletin 2022;67:71-8.
185. Wang F, Li C, Chen H, et al. Plasmonic harvesting of light energy for Suzuki coupling reactions. J Am Chem Soc 2013;135:5588-601.
186. Lanterna AE, Elhage A, Scaiano JC. Heterogeneous photocatalytic C-C coupling: mechanism of plasmon-mediated reductive dimerization of benzyl bromides by supported gold nanoparticles. Catal Sci Technol 2015;5:4336-40.
187. Li Y, Ren P, Zhang D, et al. Rationally designed metal cocatalyst for selective photosynthesis of bibenzyls via dehalogenative C-C homocoupling. ACS Catal 2021;11:4338-48.
188. Filippini G, Longobardo F, Forster L, et al. Light-driven, heterogeneous organocatalysts for C-C bond formation toward valuable perfluoroalkylated intermediates. Sci Adv 2020;6:eabc9923.
189. Zhu H, Ke X, Yang X, Sarina S, Liu H. Reduction of nitroaromatic compounds on supported gold nanoparticles by visible and ultraviolet light. Angew Chem Int Ed Engl 2010;49:9657-61.
190. Liu Z, Huang Y, Xiao Q, Zhu H. Selective reduction of nitroaromatics to azoxy compounds on supported Ag-Cu alloy nanoparticles through visible light irradiation. Green Chem 2016;18:817-25.
191. Guo X, Hao C, Jin G, Zhu HY, Guo XY. Copper nanoparticles on graphene support: an efficient photocatalyst for coupling of nitroaromatics in visible light. Angew Chem Int Ed Engl 2014;53:1973-7.
192. Brezová V, Tarábek P, Dvoranová D, Staško A, Biskupič S. EPR study of photoinduced reduction of nitroso compounds in titanium dioxide suspensions. Journal of Photochemistry and Photobiology A: Chemistry 2003;155:179-98.
193. Pal B, Torimoto T, Okazaki K, Ohtani B. Photocatalytic syntheses of azoxybenzene by visible light irradiation of silica-coated cadmium sulfide nanocomposites. Chem Commun (Camb) 2007:483-5.
194. Dai Y, Li C, Shen Y, et al. Light-tuned selective photosynthesis of azo- and azoxy-aromatics using graphitic C3N4. Nat Commun 2018;9:60.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Author Biographies





















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].