

Drug-target interaction prediction via hierarchical gated attention and information bottleneck
 0
0 Abstract
Accurate drug-target interaction (DTI) prediction is essential for drug repositioning and accelerating drug discovery. Deep learning methods have made remarkable progress over traditional biological experiments, yet existing models often fail to capture local node topologies and multi-view semantic dependencies simultaneously. Moreover, most methods rely on basic loss functions that cannot filter out redundant noise, hindering the learning of compact and discriminative node representations. In this work, we propose a DTI prediction framework that integrates a hierarchical gated multi-head attention (HGMA) mechanism with an information bottleneck (IB) strategy. HGMA adopts a two-layer architecture: the first layer performs weighted aggregation over semantic meta-paths, and the second layer fuses attention heads via an adaptive gating mechanism, enhancing drug and target representations. The IB module compresses inputs by removing task-irrelevant redundancy while preserving predictive information, improving discriminability and generalization. Extensive experiments show that our model consistently outperforms state-of-the-art methods in both accuracy and robustness.
Keywords
1. INTRODUCTION
Drug-target interaction (DTI) prediction is a fundamental task in drug discovery, facilitating the identification of potential interactions between drugs and their cognate biological targets[1]. Accurate DTI prediction provides critical insights into drug repositioning, polypharmacology, resistance mechanism analysis, and adverse effect prediction[2]. Although in vitro experimental methods for assessing DTIs are regarded as reliable, they are often expensive, time-consuming, and impractical for large-scale datasets. Consequently, computational approaches for DTI prediction have garnered increasing attention[3] owing to their ability to deliver rapid and accurate interaction predictions.
raditional computational methods fall into structure-based and ligand-based categories[4]. Structure-based methods (e.g., molecular docking) rely on three-dimensional (3D) protein structures, but their applicability is limited by the scarcity of experimentally resolved targets. Ligand-based methods assume that similar ligands exhibit similar activities, yet they depend heavily on known ligand-target interactions, which limits performance in data-sparse scenarios[5]. To address these limitations, machine learning-based approaches have been increasingly explored. Under the "guilt-by-association" principle—that similar drugs interact with similar targets—these methods infer unobserved DTIs from known interactions[6]. Machine learning methods for DTI prediction include traditional, network-based, and deep learning-based approaches. Traditional methods use handcrafted features from drug chemical structures and protein sequences, employing classifiers such as support vector machines (SVMs)[7], logistic regression (LR)[8], random forest (RF)[9], and k-nearest neighbors (KNNs)[10]. For instance, Yamanishi et al.[11] proposed a kernel-based method integrating chemical and genomic spaces for binary DTI classification, while Jacob et al.[12] introduced a multi-task SVM framework to capture inter-target relationships. Despite their successes, reliance on manual features and shallow representations limits their ability to capture complex, nonlinear relationships in high-dimensional, sparse, and heterogeneous biomedical data.
Graph-based deep learning methods have emerged as a promising paradigm for modeling biomedical network topologies. However, existing approaches face substantial challenges. First, biomedical networks are heterogeneous, encompassing diverse node and edge types that standard graph neural networks (GNNs) struggle to capture in terms of high-order semantic dependencies. Meta-paths offer multi-perspective semantics for node representations. Second, traditional graph convolutions indiscriminately aggregate neighbors, neglecting their varying importance and thereby limiting fine-grained local feature extraction. Attention mechanisms address this limitation by adaptively weighting task-relevant neighbors, enabling the simultaneous modeling of local and global dependencies. Moreover, heterogeneous networks contain noisy, redundant information. Naïve integration of multi-view features from meta-paths and attention mechanisms can yield ambiguous embeddings due to redundancy. To this end, the information bottleneck (IB) principle[13] compresses input data into compact drug-target representations, filtering noise while preserving predictive features.
Motivated by these challenges, we propose a DTI prediction framework (HGMAIB) tailored to heterogeneous biomedical networks. HGMAIB has two core components. The hierarchical gated multi-head attention (HGMA) module employs a two-tier structure: the first layer performs weighted aggregation over semantic meta-paths to capture multi-view dependencies; the second layer adaptively fuses attention head outputs via a gating mechanism, enhancing local structural representations of drugs and targets. The IB module further compresses redundant information while retaining task-relevant features, enabling the learning of compact and discriminative latent representations. The main contributions are summarized as follows:
• A HGMA mechanism is proposed to jointly model fine-grained local structural interactions and multi-view semantic information of drug and target nodes within heterogeneous biomedical networks.
• An IB module is integrated to effectively suppress redundant features and noise while preserving key predictive information, yielding compact and discriminative joint representations for DTI prediction.
• Extensive experiments conducted on two widely used benchmark DTI datasets demonstrate that the proposed HGMAIB model consistently outperforms several state-of-the-art methods.
2. RELATED WORK
2.1. Graph computational method-based DTI prediction
Early DTI prediction methods framed the task as link prediction on heterogeneous networks, learning topology-preserving embeddings. DTINet[14] integrated multimodal features via random walks followed by dimensionality reduction. With the rise of GNNs[15], NeoDTI[16] jointly modeled structural and attribute information through neighborhood aggregation, while GCN-DTI[17] and EEG-DTI[18] used graph convolutional networks (GCNs) to extract drug and protein features separately. However, these approaches rely on simplistic aggregation (e.g., mean or sum pooling), failing to capture fine-grained local structural characteristics. To better exploit semantic dependencies, researchers have introduced attention mechanisms and meta-paths. IMCHGAN[19] employed hierarchical attention to distinguish meta-path semantics, and AMGDTI[20] adaptively fused multi-view features from multiple meta-paths. DHGT-DTI[21] further proposed a dual-view heterogeneous graph to capture local and global interaction patterns. Nevertheless, naïve fusion of multi-view features often leads to information redundancy, and high-dimensional heterogeneous structures introduce noise, degrading representation compactness[22]. More recent methods adopt contrastive learning and causal inference to enhance model robustness and address data sparsity. CE-DTI[23] incorporated causal inference to mitigate bias; SGCL-DTI[24] and SHGCL-DTI[25] applied structure-aware contrastive learning to maximize mutual information between local and global views; DSS-DTI[26] used a dual-scale spatiotemporal framework; and MIDTI[27] employed multi-view interaction modeling. Despite these advances, balancing fine-grained local substructure extraction with global semantic redundancy reduction remains a key challenge, motivating a framework that integrates structural gating with information compression.
2.2. Graph attention network-based applications
The attention mechanism, inspired by human visual cognition, enables models to focus on salient input features while suppressing irrelevant information. Graph Attention Networks (GATs)[28] extend this paradigm to non-Euclidean domains. Attention mechanisms were first introduced to address long-range dependencies in machine translation. The Transformer relies entirely on self-attention to capture global dependencies, outperforming recurrent neural networks (RNNs). Attention mechanisms also capture spatial relationships and regions of interest. Non-local neural networks[29] used self-attention to model pixel-level long-range dependencies, overcoming convolution’s receptive field limits. Vision Transformers[30] further demonstrate that purely attention-based architectures excel at image recognition. Moreover, biological data (e.g., molecular structures, protein interaction networks) naturally lend themselves to graph representations. AlphaFold[31] heavily utilizes attention to predict 3D protein structures by modeling pairwise residue relationships. For molecular property prediction, GATs learn molecular fingerprints by attending to critical atoms and functional groups[32]. In protein-protein interaction (PPI) prediction, attention identifies key interface residues by weighting biologically active regions. These successes underscore the ability of graph attention mechanisms to capture both local structural details and global functional dependencies in biomedical data.
3. METHODS
3.1. Overall framework
The proposed HGMAIB model comprises four core modules [Figure 1]. (1) Node representation initialization: Node2Vec[33] generates low-dimensional embeddings for drug and protein nodes as informative input features; (2) Adaptive meta-path selection: Automatically identifies discriminative semantic paths to guide high-order semantic information propagation and aggregation; (3) Multi-semantic structural modeling: Employs a multi-step graph convolutional framework with weighted residual connections to capture deep semantic dependencies while mitigating gradient vanishing and feature over-smoothing. A hierarchical gated multi-head attention mechanism further learns node-level local structural patterns and integrates multi-perspective semantic information. (4) IB: Compresses redundant information and extracts task-relevant joint embeddings, enhancing discriminative power and generalization.
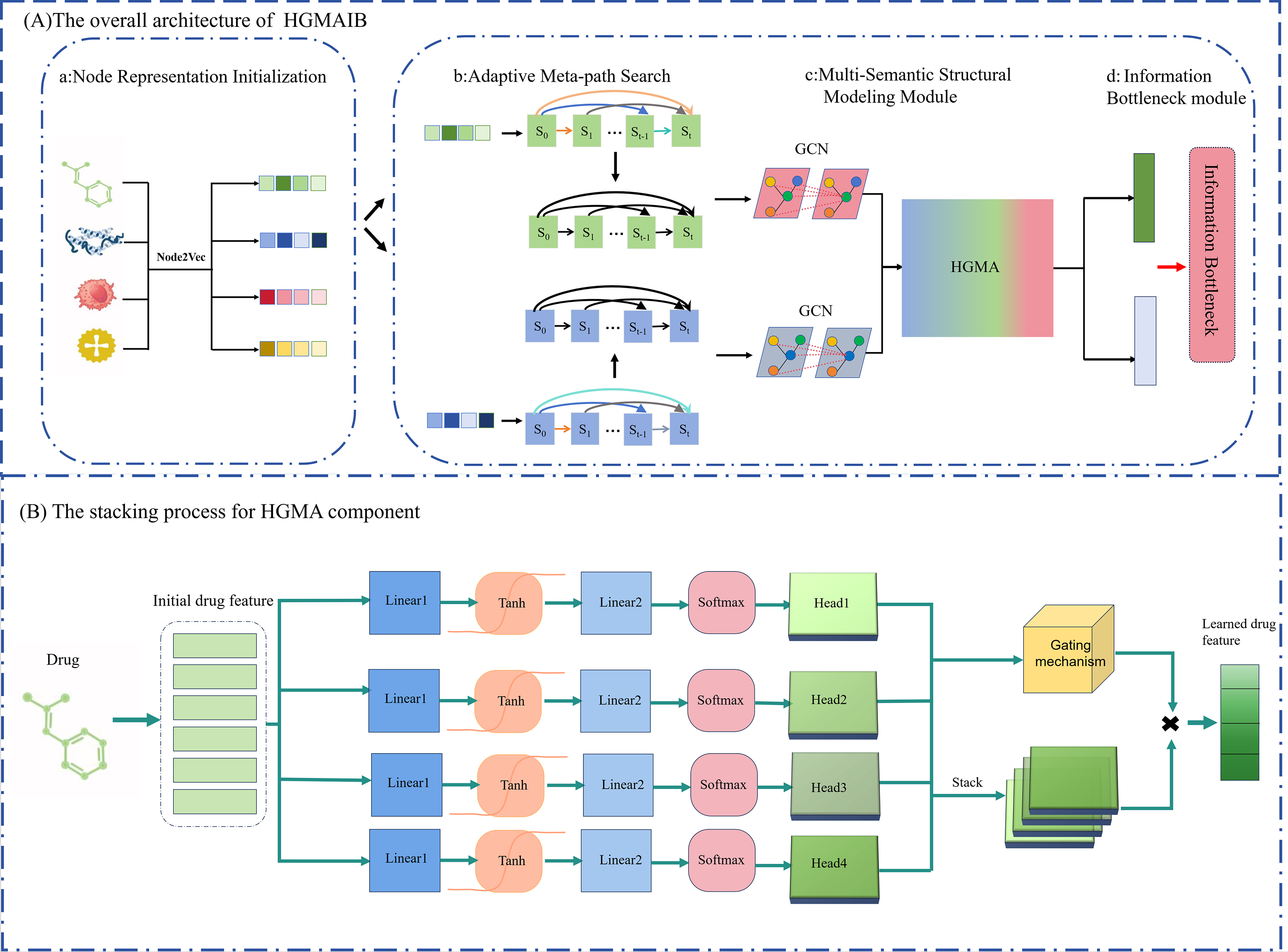
Figure 1. (A) Overall architecture of HGMAIB: (a) Node representation initialization - Node2Vec extracts drug and target features. (b) Adaptive meta-path search - A dynamic search strategy identifies informative semantic paths. (c) Multi-semantic structural modeling - Multi-step residual graph convolution and hierarchical gated multi-head attention (HGMA) capture deep dependencies. (d) Information bottleneck - Learned embeddings are refined to filter redundant noise. (B) HGMA stacking process: The hierarchical architecture performs weighted aggregation within each attention head, followed by a gating mechanism that integrates information across all heads.
3.2. Node representation initialization module
By simulating biased random walks, Node2Vec captures both local and global structural information and generates low-dimensional dense vectors for each node, serving as input features for downstream GNNs [Figure 2]. First, an undirected weighted bipartite graph is constructed from the drug-target adjacency matrix, where drugs (D1-D4) and targets (T1-T4) form two distinct node types, and edge weights denote known interactions. To prevent data leakage, the bipartite graph used for Node2Vec initialization is dynamically rebuilt during each five-fold cross-validation iteration: all test-set edges are explicitly masked and excluded from the adjacency matrix before random walk generation. Node2Vec is then applied to the masked graph to obtain initial node embeddings. It then simulates fixedlength random walks by interpolating between breadth first search (BFS) and depth first search (DFS). Two hyperparameters control walk preferences: the return parameter p (the likelihood of revisiting the previous node) and the inout parameter q (the tendency to explore local vs. distant nodes), thereby balancing structural equivalence and homophily.
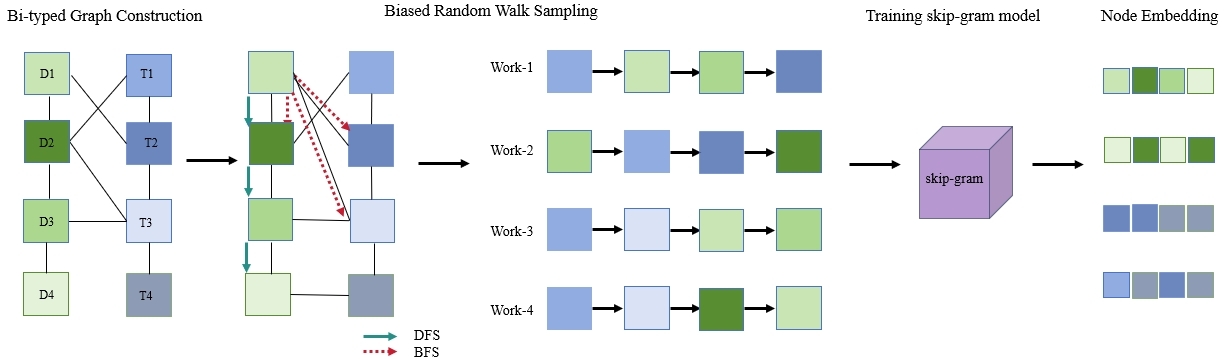
Figure 2. Workflow of Node2Vec for node representation learning.
The resulting walk sequences are treated as “sentences” and fed into a skip-gram model[34]. Originally designed for natural language processing (NLP), the skip-gram model maximizes word co-occurrence probabilities; in graphs, it captures the co-occurrence of adjacent nodes within the same walk, encoding contextual structure. After training, each node is mapped to a dense low-dimensional embedding that preserves the drug-target graph topology. These embeddings serve as informative initial inputs for subsequent GNNs, enhancing their ability to perceive both topological and semantic features.
3.3. Adaptive metapath selection module
Inspired by AMGDTI[20], we adopt a dynamic search strategy to model high-order semantic dependencies in heterogeneous graphs. This mechanism automatically generates discriminative meta-paths, constructing a guided graph structure that enhances semantic propagation and feature aggregation. Its key advantage is eliminating manual path design by adaptively identifying optimal meta-path combinations, thereby improving the model’s ability to represent complex semantic relationships.
Specifically, this module constructs a directed acyclic adaptive meta-graph, denoted as M = (Vm, Em). The node set Vm represents the sequence of node feature states {S0, S1, …, ST} after each propagation step within the heterogeneous network, where the total number of nodes is determined by the predefined propagation steps T. The edge set Em encodes all possible information propagation strategies. For example, a directed edge labeled "Protein→Disease" from S0 to S1 indicates that the feature of a "Disease" node in S1 is obtained by aggregating the features of "Protein" nodes in S0. This structure allows any previous state Si∈{S0, S1,…, ST-1} to influence the current state ST through skip connections, enabling the model to fully capture complex semantic information embedded in heterogeneous networks.
In addition, the model adaptively determines each edge in the meta-graph by evaluating whether a previous state contributes to the current state and selecting the most appropriate propagation strategy. This approach treats all edge types as potential propagation patterns, enabling dynamic path selection. Additionally, two auxiliary connection types are introduced: L1 indicates that the current state is identical to the previous state, while L2 implies that the previous state does not affect the current state. The adaptive meta-graph comprises 12 possible edge types: {LDP, LPD, LDS, LSD, LDE, LED, LPE, LEP, LDD, LPP, L1, L2}. The first ten correspond to explicit biological relations within the heterogeneous network (e.g., LDP: Drug → Protein, LED: Disease → Drug), while the latter two are auxiliary designs intended to enhance the structural flexibility and semantic expressiveness of the model.
To mathematically formulate the path-selection optimization and ensure reproducibility, we formally define the candidate connection constraints and the selection mechanism. Let Ct,i denote the set of possible connection types from a previous state Si to the current state St. To avoid meaningless aggregations, Ct,i is dynamically constrained based on the propagation step t and the total steps T:
where EM represents the full set of 12 candidate edges, and R represents the subset of target-related edges. Subsequently, we assign a learnable structural parameter
where m is the index of the edge type with the maximum parameter value (
3.4 Multi-Semantic Structural Modeling Module
(1) Multi-step Weighted residual graph convolution module
This module aims to capture the topological information and structural dependencies of nodes along different semantic paths by integrating multi-hop graph propagation with residual learning. It enhances the expressive power of node features while effectively mitigating issues such as gradient vanishing and over-smoothing during deep propagation. Given the input feature matrix
where
where
where
The final node representation at step t is obtained by summing the sequential propagation
After T propagation steps, the final node representation is obtained as
where σ is the Sigmoid Linear Unit (SiLu) activation function[35], BN denotes batch normalization[36], and
(2) Hierarchical gated multi-head attention module
To capture local structural dependencies and integrate multi-view semantic information, the HGMA module uses a hierarchical gated multi-head attention mechanism with two layers. The first layer performs weighted aggregation along distinct semantic paths within each attention head. The second layer then fuses information across heads via a gating mechanism. Concretely, after multi-path residual GCN propagation, the representations of each node across all semantic paths are collected. The final representations from all L paths are stacked along the path dimension into a 3D tensor as follows:
where
where
where
where ⊙ denotes element-wise multiplication, and hl denotes the node representations under the l-th semantic path obtained from Eq. (6). After the first-level attention, each attention head h = {1,…,M} outputs an aggregated node representation o(h). The representations are then stacked by the formula:
To further regulate the contribution of each attention head to the final representation, a gating mechanism[39] is introduced to adaptively weight and fuse the outputs of all attention heads. Specifically, mean pooling is first applied along the feature dimension d to each attention head’s output to obtain an average activation value, reflecting the overall contribution of each head to the node representation. This value is then mapped to the range [0,1] via a sigmoid function[37], producing the gating weights G as follows:
where σ(•) denotes the sigmoid function, which maps the input logit to a probability score in the range (0, 1). Finally, the gating weights G are multiplied element-wise with the attention head outputs, and summed along the head dimension to obtain the final fused representation as follows:
where ⊙ denotes element-wise multiplication, and Hfinal denotes the final fused node representation obtained by aggregating the outputs of all attention heads through a learnable gating mechanism.
3.5. Information bottleneck module
To learn compact and discriminative drug-target representations, we integrate the IB principle[13,40] into our framework. IB extracts target-relevant information from inputs by balancing compression and prediction. We adopt a variational implementation, modeling each node representation as a latent probabilistic distribution. Variational inference approximates the posterior, and a Kullback-Leibler (KL) divergence term[41] regularizes it toward a standard normal prior. This mechanism retains discriminative features and filters redundancy. It guides the model to learn robust and compact embeddings. Specifically, for each drug and target node, the encoder outputs the mean μ and log-variance logσ2 of the latent representation, which parameterize a Gaussian distribution as follows:
where diag denotes the operation of converting a vector into a diagonal matrix. Since direct sampling from this distribution does not support gradient backpropagation, the reparameterization trick[42] is employed to enable differentiable sampling of the latent variables:
where ε denotes a random variable sampled from the standard normal distribution, and ⊙ represents element-wise multiplication. The sampled drug and target representations, Zs and Zt, are then used to compute the interaction prediction score, defined as their inner product:
The prediction error is measured using the Binary Cross-Entropy (BCE) loss[43] between the predicted interaction score and the ground truth label:
where yi is the ground truth label, si is the predicted interaction score, N is the total number of training samples, and σ is the sigmoid function. To regulate the degree of information compression, the KL divergence is introduced as a regularization term, encouraging the latent variable distribution to approximate a unit Gaussian:
The final loss function is formulated as:
where LBCE is the BCE loss, β is a hyperparameter controlling the strength of the IB regularization,
4. EXPERIMENTAL SETUP
4.1. Datasets
To evaluate the proposed method, we used two public benchmark datasets: the Luo dataset[14] and the Zheng dataset[44]. The Luo dataset comprises four biomedical entity types (drugs, proteins, diseases, side effects) and multiple relation types (e.g., drug-target, disease-protein, drug-side effect), offering a heterogeneous network with diverse structural information [Table 1]. The Zheng dataset provides rich attribute features—chemical substructures and substituents for drugs, and Gene Ontology (GO) terms for proteins—along with associated edges (e.g., drug-substructure, protein-GO), enabling fine-grained semantic modeling [Table 2].
Information on nodes and edges in the Luo dataset
| Node type | Num | Edge type | Num |
| Drug | 708 | Drug-drug (interaction) | 10036 |
| Protein | 1512 | Drug-drug (similarity) | 501264 |
| Disease | 5603 | Drug-protein | 1923 |
| Side effect | 4192 | Drug-disease | 199214 |
| Drug-side effect | 80164 | ||
| Protein-disease | 1596745 | ||
| Protein-protein (interaction) | 7363 | ||
| Protein-protein (similarity) | 2286144 |
Information on nodes and edges in the Zheng dataset
| Node type | Num | Edge type | Num |
| Drug | 1094 | Drug-drug | 1196836 |
| Protein | 1556 | Drug-protein | 11819 |
| Chemical structure | 881 | Drug-chemical substructure | 133880 |
| Side effect | 4063 | Drug-side effect | 122792 |
| Substituent | 738 | Drug-substituent | 20798 |
| GO term | 4098 | Protein-GO term | 35980 |
| Protein-protein | 2421136 |
4.2. Experimental settings
For reproducibility, all code and datasets are available at https://github.com/chenzh-23/HGMAIB. The models were developed with Python 3.8, PyTorch 1.11+cu113, and SciPy 1.10.1. Data are presented as
A fivefold crossvalidation strategy was adopted. In each fold, 60% of the samples were used for training, 20% for validation (hyperparameter tuning), and 20% for testing. To address the severe imbalance between known and unknown interactions, negative samples were randomly undersampled to match positive samples per fold. Two standard evaluation metrics widely used in previous studies for assessing binary classification models in DTI prediction were adopted: the Area Under the Receiver Operating Characteristic Curve (AUC)[46] and the Area Under the Precision-Recall Curve (AUPRC)[47].
5. RESULTS AND DISCUSSION
5.1. Comparison with baselines
To comprehensively evaluate HGMAIB, we compare it with twelve baseline methods spanning diverse modeling paradigms. For a fair comparison, architectural hyperparameters (e.g., number of layers, embedding dimensions) follow each baseline’s original optimal settings. Training hyperparameters (learning rate, weight decay, early stopping patience) were systematically retuned for each baseline on the Luo and Zheng datasets using validation set performance within our five-fold cross-validation framework. The baselines include:
• Network-based DTI methods: DTINet[14] and NeoDTI[16].
• GNN-based DTI models: GCN-DTI[17], EEG-DTI[18], and IMCHGAN[19].
• Representation enhancement and contrastive learning-based methods: CE-DTI[23], SGCL-DTI[24], MIDTI[27], and SHGCL-DTI[25].
• Recent state-of-the-art heterogeneous graph frameworks: AMGDTI[20], DSS-DTI[26], and DHGT-DTI[21].
As shown in Table 3, HGMAIB consistently achieves the best performance across all evaluation metrics on both benchmark datasets, significantly outperforming all baseline methods. Specifically, HGMAIB attains values of 0.991 ± 0.002 and 0.990 ± 0.002 on the Luo dataset, and 0.988 ± 0.002 and 0.984 ± 0.001 on the Zheng dataset, demonstrating its superior predictive accuracy and robustness.
Performance comparison with baseline methods on the Luo and Zheng datasets
| Methods | Luo dataset | Zheng dataset | ||
| AUC | AUPRC | AUC | AUPRC | |
| DTINet | 0.879 ± 0.004** | 0.906 ± 0.003** | 0.889 ± 0.004** | 0.900 ± 0.004** |
| NeoDTI | 0.955 ± 0.003** | 0.889 ± 0.004** | 0.946 ± 0.003** | 0.846 ± 0.005** |
| GCN-DTI | 0.918 ± 0.005** | 0.897 ± 0.005** | 0.922 ± 0.004** | 0.914 ± 0.004** |
| IMCHGAN | 0.956 ± 0.004** | 0.959 ± 0.003** | 0.946 ± 0.002** | 0.929 ± 0.003** |
| SGCL-DTI | 0.977 ± 0.002** | 0.976 ± 0.002** | 0.942 ± 0.003** | 0.941 ± 0.003** |
| EEG-DTI | 0.954 ± 0.003** | 0.964 ± 0.004** | 0.968 ± 0.002** | 0.968 ± 0.002** |
| MIDTI | 0.978 ± 0.003** | 0.970 ± 0.002** | 0.957 ± 0.003** | 0.961 ± 0.003** |
| CE-DTI | 0.976 ± 0.002** | 0.976 ± 0.002** | 0.972 ± 0.003** | 0.972 ± 0.002** |
| SHGCL-DTI | 0.957 ± 0.004** | 0.958 ± 0.003** | 0.954 ± 0.002** | 0.949 ± 0.004** |
| DSS-DTI | 0.986 ± 0.003* | 0.985 ± 0.002* | 0.972 ± 0.001** | 0.969 ± 0.002** |
| DHGT-DTI | 0.965 ± 0.003** | 0.969 ± 0.001** | 0.973 ± 0.002** | 0.977 ± 0.001** |
| AMGDTI | 0.977 ± 0.002** | 0.977 ± 0.002** | 0.973 ± 0.004** | 0.971 ± 0.002** |
| HGMAIB | 0.991 ± 0.002 | 0.990 ± 0.002 | 0.988 ± 0.002 | 0.984 ± 0.001 |
Compared with early network-based methods (DTINet, NeoDTI), HGMAIB achieves substantial gains, revealing the limitations of shallow feature projection and simple network integration. Against GNN-based models (GCN-DTI, IMCHGAN), HGMAIB improves performance by explicitly modeling heterogeneous semantic paths rather than relying solely on homogeneous aggregation. It also surpasses contrastive learning methods (CE-DTI, SGCL-DTI, MIDTI, SHGCL-DTI), which lack redundancy suppression; in contrast, HGMAIB’s information bottleneck retains task-relevant features, yielding more discriminative representations. Among recent heterogeneous frameworks, DSS-DTI and DHGT-DTI capture multi-scale or dual-view dependencies, but HGMAIB further integrates hierarchical gated multi-head attention with IB-guided refinement, jointly modeling multi-semantic structures for more robust and accurate DTI prediction. Collectively, these results demonstrate HGMAIB’s ability to capture complex heterogeneous semantics and generate highly discriminative representations.
5.2. Ablation study
To validate the contribution of each component, we designed four ablation experiments targeting the multi-step weighted residual graph convolution, HGMA, IB, and adaptive meta-path search. The results are shown in Figure 3.
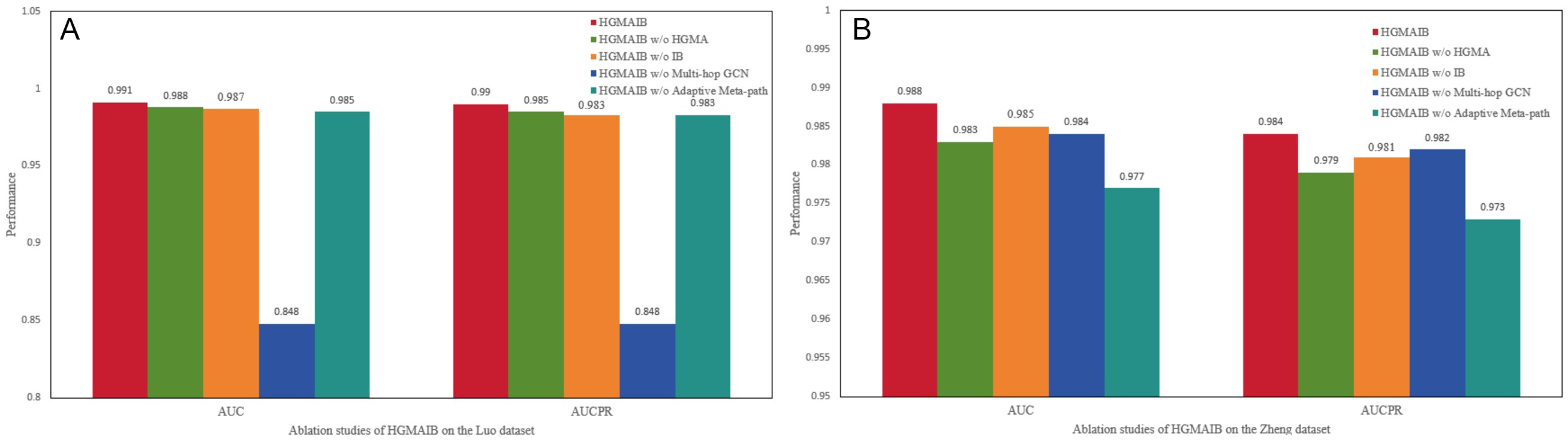
Figure 3. Performance comparison of ablation experiments: (A) Results on the Luo dataset; (B) results on the Zheng dataset.
• HGMAIB w/o HGMA: This variant replaces the HGMA module with a single-head attention mechanism, while keeping the remaining architecture unchanged.
• HGMAIB w/o IB: In this variant, the IB loss is substituted with the conventional BCE loss, and all other modules are preserved.
• HGMAIB w/o Multi-hop GCN: This variant replaces the multi-step graph convolution module with a basic first-order graph convolution, removing residual connections and deep structural propagation, with the rest of the framework kept intact.
• HGMAIB w/o Adaptive Meta-path: This variant removes the adaptive meta-path search module and instead adopts a fixed predefined meta-path, namely Drug → Disease → Protein → Protein, as the structural input for message propagation, while keeping all other components unchanged.
The ablation results confirm that removing any component degrades performance. On the Luo dataset, replacing hierarchical gated multi-head attention with single-head attention lowers the AUC to 0.988 and the AUPRC to 0.985, highlighting the importance of multi-head attention with hierarchical gating for multi-semantic aggregation. Replacing the IB loss with standard BCE loss yields a slight performance decrease (AUC 0.987, AUPRC 0.984), validating IB’s role in suppressing redundancy. Removing the adaptive meta-path selection and adopting a fixed semantic path (Drug → Disease → Protein → Protein) further degrades performance (AUC 0.985, AUPRC 0.983). Replacing the multi-step graph convolution and residual connections with a single-layer convolution causes a sharp performance drop on the sparse Luo dataset (AUC and AUPRC both fall to 0.848). With only 1,923 edges among 708 drugs and 1,512 proteins (density ≈ 0.18%), deep multi-hop propagation and residual learning are essential to capture higher-order dependencies. In contrast, on the denser Zheng dataset (11,819 edges, density ≈ 0.69%, nearly four times denser than Luo), the performance loss is much milder (AUC 0.977, AUPRC 0.973). The Zheng dataset contains rich local attributes (881 chemical substructures, 4,098 GO terms), which provide sufficient 1-hop semantic signals, partly alleviating the need for deep propagation. These results confirm that each component of HGMAIB is indispensable, and that the benefit of deep structural propagation depends on the inherent complexity of the biomedical network.
5.3. Effect of node embedding methods
To justify the use of Node2Vec for node initialization in HGMAIB, we compared it with LINE and GraphSAGE on the Luo dataset. All other components (network architecture, training procedure, hyperparameters) were kept identical. As shown in Table 4, Node2Vec achieves the highest predictive performance, outperforming both alternatives. This indicates that Node2Vec more effectively captures the structural and semantic information of heterogeneous nodes in biomedical networks, which is essential for accurate DTI prediction.
Effect of node embedding methods on HGMAIB performance on the Luo dataset
| Node embedding method | AUC | AUPRC |
| Node2Vec | 0.991 | 0.990 |
| LINE | 0.982 | 0.976 |
| GraphSAGE | 0.980 | 0.968 |
5.4. Parameter sensitivity analysis
Parameter sensitivity analysis was conducted on the Luo dataset to evaluate the impact of three key hyperparameters: the number of attention heads, learning rate, and the β coefficient. The number of attention heads (tested in {2, 4, 8, 16}) achieved peak performance with four heads [Figure 4A], indicating that a moderate size optimally balances feature aggregation and computational efficiency. The learning rate (tested in {5e-4, 1e-3, 5e-3, 1e-2}) yielded the best results at 5e-3 [Figure 4B], balancing convergence speed and stability—too small a value slows training, while too large a value causes instability.
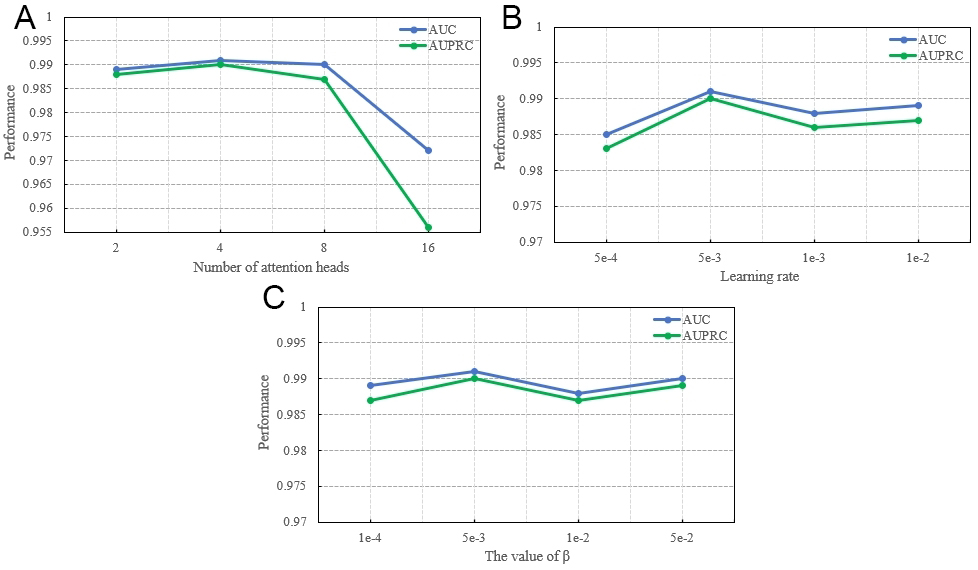
Figure 4. The performance of HGMAIB on the Luo dataset under different parameter settings: (A) Number of attention heads; (B) Learning rate; (C) β coefficient.
The β parameter determines the weight of the IB loss and plays a key role in balancing representation learning and regularization. A small β may lead to under-regularization and overfitting, whereas an excessively large β can constrain the model and impair learning. β was evaluated over the set {1e-3, 5e-3, 1e-2, 5e-2}, with optimal performance observed at 5e-3, achieving a favorable trade-off between effective representation learning and appropriate regularization [Figure 4C].
5.5. t-SNE visualization of node embeddings
To evaluate HGMAIB’s representation capability, t-SNE was applied to the Zheng dataset, projecting drug (blue) and target (red) embeddings into a two-dimensional space. As shown in Figure 5A, the initial embeddings exhibit substantial overlap with no distinct clusters, indicating limited semantic discriminability. After training [Figure 5B], the embeddings form compact intra-class clusters with clear inter-class separation and well-defined boundaries. This improvement demonstrates that HGMAIB effectively captures heterogeneous semantic dependencies and topological correlations: the hierarchical gated multi-head attention adaptively aggregates multi-level semantics, while the IB filters redundant signals and preserves task-relevant features. Consequently, HGMAIB produces discriminative, biologically meaningful embeddings, enhancing interpretability and generalization in DTI prediction. Overall, these results confirm HGMAIB’s ability to learn robust representations from complex heterogeneous graphs.
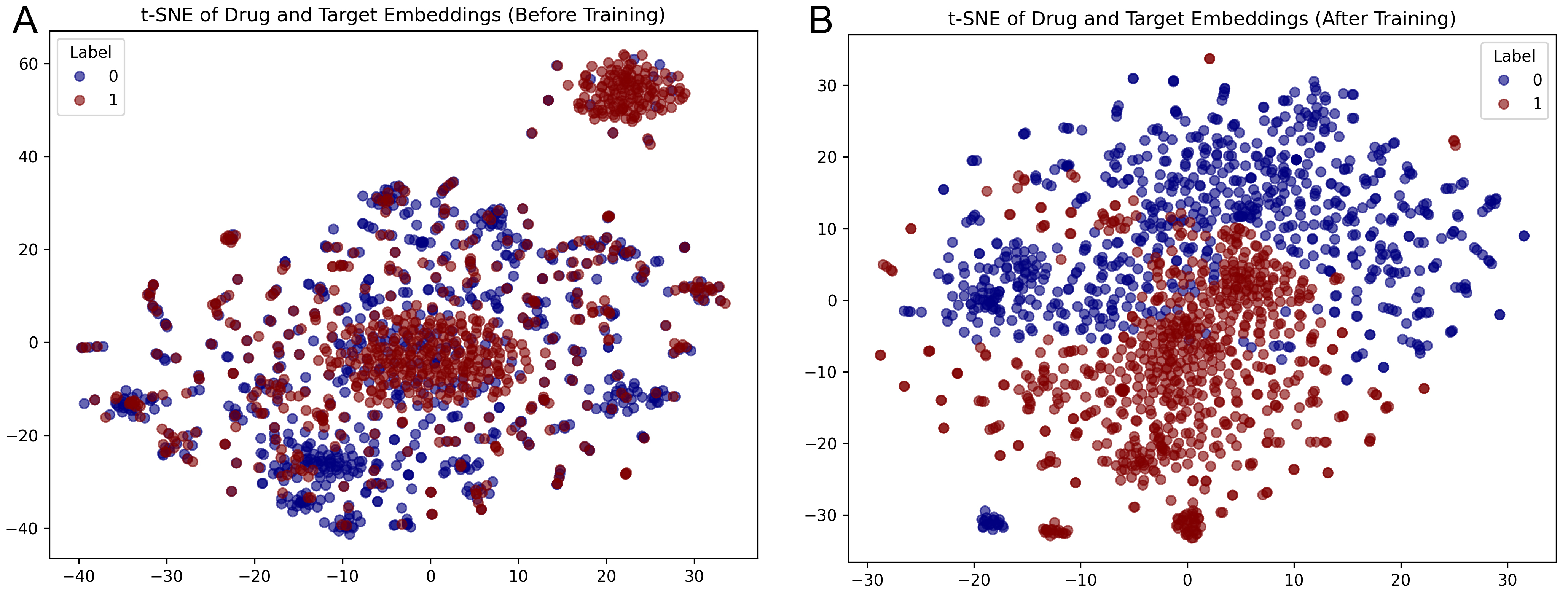
Figure 5. t-SNE visualization of drug and target embeddings on the Zheng dataset. (A) Initial embeddings before training; (B) Learned embeddings after training with the HGMAIB model.
5.6. Computational cost analysis
To improve the completeness of our evaluation and justify the computational cost of the proposed complex modules against baseline methods, we provide a detailed theoretical complexity analysis and an empirical footprint of HGMAIB. Because existing baseline methods are implemented across varied frameworks, making direct empirical runtime comparisons highly hardware-dependent, we focus on asymptotic complexity and actual resource consumption.
The computational complexity of HGMAIB is primarily composed of three phases. In the multi-path graph representation learning phase, multi-step sequential propagation with residual aggregation over P meta-paths incurs a complexity of O(T2 × |E| × D + P × N × D), where N is the number of nodes, |E| is the number of edges, D is the hidden dimension, and T is the propagation steps. In the hierarchical attention aggregation phase, path-level and head-level gated attention across M heads contributes O(N × P × D × M + N × M × D). Finally, the IB-based feature compression requires O(N × D2) operations. Consequently, the overall time complexity scales linearly with the number of edges and attention heads, avoiding exponential computational overhead. The space complexity (parameter size and feature storage) is strictly bounded by O(|E|+N × D + P × N × D + N × P × M).
Empirically, to further evaluate the practical efficiency of HGMAIB against baseline scales, we recorded the computational resource consumption during training. On the Luo dataset, each training fold processes approximately 2,300 meta-path-guided subgraph instances (averaging 3,570 nodes and 8.1 million edges per subgraph). Evaluated on a single NVIDIA RTX 3090 GPU, HGMAIB achieves stable training with an average training time of approximately 15 seconds per fold and a remarkably low peak GPU memory usage of 0.38 GB. These empirical results clearly indicate that, despite incorporating advanced modules such as multi-step residual propagation and hierarchical multi-path attention, the parameter footprint and computational overhead of HGMAIB remain highly competitive with basic GNNs. This demonstrates a highly favorable and justifiable trade-off between complex model expressiveness and computational cost in large-scale heterogeneous biomedical networks.
6. CONCLUSION
To address the critical challenge of accurate DTI identification in drug repositioning, this study proposes a novel predictive framework termed HGMAIB. Extensive evaluations demonstrate that HGMAIB achieves highly competitive predictive performance by effectively capturing complex structural dependencies and fusing multi-perspective semantic information within heterogeneous biological networks. Furthermore, our findings confirm that the synergistic integration of the HGMA and the IB module critically enhances overall model efficacy by actively filtering out redundant noise to produce highly discriminative representations. Future research will focus on incorporating broader heterogeneous biomedical data and dynamic GNN techniques[48] to capture temporal biological dynamics, thereby further advancing robust DTI prediction and accelerating drug discovery.
DECLARATIONS
Authors’ contributions
Substantial contributions to the conceptualization, methodology, writing, and visualization: Song, S.;
Formal analysis, methodology, and validation, along with data analysis: Wang, Y.; Guo, Q.
Availability of data and materials
The experimental dataset supporting this study can be obtained from
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Grant No. 62403437) and the Young Backbone Teacher Training Program of Zhengzhou University of Light Industry (Grant No. 13502010009).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Zhou, G.; Xuan, C.; Wang, Y.; Zhang, B.; Wu, H.; Gao, J. Drug repositioning based on a multiplex network by integrating disease, gene, and drug information. Curr. Bioinf. 2023, 18, 266-75.
2. Palhamkhani, F.; Alipour, M.; Dehnad, A.; Abbasi, K.; Razzaghi, P.; Ghasemi, J. B. DeepCompoundNet: enhancing compound-protein interaction prediction with multimodal convolutional neural networks. J. Biomol. Struct. Dyn. 2023, 43, 1414-23.
3. Hu, L.; Fu, C.; Ren, Z.; et al. SSELM-neg: spherical search-based extreme learning machine for drug-target interaction prediction. BMC. Bioin. Bioinform. 2023, 24, 38.
4. Shi, W.; Yang, H.; Xie, L.; Yin, X.; Zhang, Y. A review of machine learning-based methods for predicting drug-target interactions. Health. Inf. Sci. Syst. 2024, 12, 30.
5. Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M. M. Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. Inf. Fusion. 2019, 50, 71-91.
6. Zhang, R.; Wang, Z.; Wang, X.; Meng, Z.; Cui, W. MHTAN-DTI: metapath-based hierarchical transformer and attention network for drug-target interaction prediction. Brief. Bioinform. 2023, 24, bbad079.
7. Yang, Y.; Yang, Y.; Pan, A.; et al. Identifying depression in Parkinson's disease by using combined diffusion tensor imaging and support vector machine. Front. Neurol. 2022, 13, 878691.
8. Yang, J.; He, S.; Zhang, Z.; Bo, X. NegStacking: drug-target interaction prediction based on ensemble learning and logistic regression. IEEE/ACM. Trans. Comput. Biol. Bioinform. 2021, 18, 2624-34.
9. De, A.; Chowdhury, A. S. DTI based Alzheimer’s disease classification with rank modulated fusion of CNNs and random forest. Expert. Syst. Appl. 2021, 169, 114338.
10. Liu, B.; Pliakos, K.; Vens, C.; Tsoumakas, G. Drug-target interaction prediction via an ensemble of weighted nearest neighbors with interaction recovery. Appl. Intell. 2021, 52, 3705-27.
11. Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232-40.
12. Jacob, L.; Vert, J. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics 2008, 24, 2149-56.
13. Tishby, N.; Pereira, F. C.; Bialek, W. The information bottleneck method. arXiv 2000, arXiv:physics/0004057.
14. Luo, Y.; Zhao, X.; Zhou, J.; et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573.
15. Scarselli, F.; Gori, M.; Tsoi, A. C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE. Trans. Neural. Netw. 2009, 20, 61-80.
16. Wan, F.; Hong, L.; Xiao, A.; Jiang, T.; Zeng, J. NeoDTI: neural integration of neighbor information from a heterogeneous network for discovering new drug-target interactions. Bioinformatics 2019, 35, 104-11.
17. Zhao, T.; Hu, Y.; Valsdottir, L. R.; Zang, T.; Peng, J. Identifying drug-target interactions based on graph convolutional network and deep neural network. Brief. Bioinform. 2021, 22, 2141-50.
18. Peng, J.; Wang, Y.; Guan, J.; et al. An end-to-end heterogeneous graph representation learning-based framework for drug-target interaction prediction. Brief. Bioinform. 2021, 22, bbaa430.
19. Li, J.; Wang, J.; Lv, H.; Zhang, Z.; Wang, Z. IMCHGAN: inductive matrix completion with heterogeneous graph attention networks for drug-target interactions prediction. IEEE/ACM. Trans. Comput. Biol. Bioinform. 2022, 19, 655-65.
20. Su, Y.; Hu, Z.; Wang, F.; et al. AMGDTI: drug-target interaction prediction based on adaptive meta-graph learning in heterogeneous network. Brief. Bioinform. 2024, 25, bbad474.
21. Wang, M.; Lei, X.; Guo, L.; Chen, M.; Pan, Y. DHGT-DTI: advancing drug-target interaction prediction through a dual-view heterogeneous network with GraphSAGE and graph transformer. J. Pharm. Anal. 2025, 15, 101336.
22. Zhang, Z.; Zhou, X.; Qi, Y.; et al. Leveraging 3D molecular spatial visual information and multi-perspective representations for drug discovery. Adv. Sci. 2025, 13, e12453.
23. Qiao, G.; Wang, G.; Li, Y. Causal enhanced drug-target interaction prediction based on graph generation and multi-source information fusion. Bioinformatics 2024, 40, btae570.
24. Li, Y.; Qiao, G.; Gao, X.; Wang, G. Supervised graph co-contrastive learning for drug-target interaction prediction. Bioinformatics 2022, 38, 2847-54.
25. Yao, K.; Wang, X.; Li, W.; et al. Semi-supervised heterogeneous graph contrastive learning for drug-target interaction prediction. Comput. Biol. Med. 2023, 163, 107199.
26. Wu, M.; Guo, C.; Ning, Q.; Li, H.; Guo, S.; Deng, Z. Dss-Dti: drug-target interaction prediction method based on dual spatiotemporal scales. SSRN 2025.
27. Song, W.; Xu, L.; Han, C.; Tian, Z.; Zou, Q. Drug-target interaction predictions with multi-view similarity network fusion strategy and deep interactive attention mechanism. Bioinformatics 2024, 40, btae346.
28. Velickovic, P.; Cucurull, G.; Casanova, A.; et al. Graph attention networks. arXiv 2017, abs/1710.10903.
29. Wang, X.; Girshick, R.; Gupta, A.; He, K. Non-local neural networks. In 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR); 2018 Jun 18-23; Salt Lake City, UT, USA. IEEE; 2018. pp. 7794-803.
30. Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; et al. An image is worth 16x16 words: transformers for image recognition at scale. ArXiv 2020, abs/2010.11929.
31. Jumper, J.; Evans, R.; Pritzel, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583-9.
32. Xiong, Z.; Wang, D.; Liu, X.; et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2019, 63, 8749-60.
33. Balvir, S. U.; Raghuwanshi, M. M.; Borkar, P. S. Node2Vec and machine learning: a powerful duo for link prediction in social network. J. Electr. Syst. 2024, 20, 639-49.
34. Mikolov, T.; Sutskever, I.; Chen, K.; et al. Distributed representations of words and phrases and their compositionality. Adv. Neural. Inform. Process. Syst. 2013, arXiv:1310.4546.
35. Singh, B.; Patel, S.; Vijayvargiya, A.; Kumar, R. Analyzing the impact of activation functions on the performance of the data-driven gait model. Results. Eng. 2023, 18, 101029.
37. Dubey, S. R.; Singh, S. K.; Chaudhuri, B. B. Activation functions in deep learning: a comprehensive survey and benchmark. Neurocomputing 2022, 503, 92-108.
38. Shen, K.; Guo, J.; Tan, X.; et al. A study on relu and softmax in transformer. arXiv 2023, arXiv:2302.06461.
39. Du, Y.; Liu, Y.; Peng, Z.; Jin, X. Gated attention fusion network for multimodal sentiment classification. Knowl. Based. Syst. 2022, 240, 108107.
40. Sun, Q.; Li, J.; Peng, H.; et al. Graph structure learning with variational information bottleneck. AAAI 2022, 36, 4165-74.
41. Wu, T.; Tao, C.; Wang, J.; et al. Rethinking kullback-leibler divergence in knowledge distillation for large language models. arXiv 2024, arXiv:2404.02657.
42. Huang, W.; Chang, W.; Yan, G.; Yang, Z.; Luo, H.; Pei, H. EEG-based motor imagery classification using convolutional neural networks with local reparameterization trick. Expert. Syst. Appl. 2022, 187, 115968.
43. Tian, Y.; Wang, X.; Yao, X.; Liu, H.; Yang, Y. Predicting molecular properties based on the interpretable graph neural network with multistep focus mechanism. Brief. Bioinform. 2023, 24, bbac534.
44. Zheng, Y.; Peng, H.; Zhang, X.; Gao, X.; Li, J. Predicting drug targets from heterogeneous spaces using anchor graph hashing and ensemble learning. In 2018 International Joint Conference on Neural Networks (IJCNN); 2018 Jul 8-13; Rio de Janeiro. IEEE; 2018. pp. 1-7.
45. Fan, L.; Long, Z. Optimization of Nadam algorithm for image denoising based on convolutional neural network. In 2020 7th International Conference on Information Science and Control Engineering (ICISCE); 2020 Dec 18-20; Changsha, China. IEEE; 2020. pp. 957-61.
46. Carrington, A. M.; Manuel, D. G.; Fieguth, P. W.; et al. Deep ROC analysis and AUC as balanced average accuracy, for improved classifier selection, audit and explanation. IEEE. Trans. Pattern. Anal. Mach. Intell. 2023, 45, 329-41.
47. Lai, B.; Xu, J. Accurate protein function prediction via graph attention networks with predicted structure information. Brief. Bioinform. 2022, 23, bbab502.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].