
Targeting glial dysfunction in Alzheimer’s disease: insights into pathogenesis and emerging therapeutics
 0
0 
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive decline, amyloid-β (Aβ) plaques, and neurofibrillary tangles. Despite extensive research, its pathogenesis remains incompletely understood and no disease-modifying therapies are currently available. While early studies focused on neuronal dysfunction, growing evidence implicates glial cells - including microglia, astrocytes, and oligodendrocytes - in driving AD pathogenesis. Microglia initially clear Aβ and cellular debris; however, chronic activation triggers the release of proinflammatory cytokines that worsen Aβ and tau pathology and induce neurotoxic astrocytes. Reactive astrocytes compromise blood-brain barrier integrity, secrete inflammatory mediators, and impair synaptic transmission. Oligodendrocytes and their progenitors, beyond their role in myelination, can adopt disease-associated states that alter metabolic support and immune signaling, yielding both protective and detrimental effects. Recent multi-omics studies have identified critical regulatory pathways, including NF-κB, the NLRP3 inflammasome, and the cGAS-STING axis, that govern glial phenotype transitions. Therapeutic strategies targeting these pathways, such as small-molecule inhibitors, immunomodulators, and NAD+ precursors, have shown promise in preclinical AD models by reducing neuroinflammation, restoring glial homeostasis, and improving cognition. In this review, we summarize glial cell functions in health and disease, dissect molecular mechanisms of glial dysfunction in AD, and evaluate emerging glia-directed therapies. Finally, we discuss translational challenges and outline future directions for leveraging glial biology in the development of effective AD therapies.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia worldwide, accounting for approximately 70% of all cases, and poses a substantial healthcare burden due to its progressive and inexorable decline in cognitive function[1,2]. Clinically, AD manifests as deficits in memory, language, executive function, and visuospatial skills, ultimately compromising the ability to perform routine daily activities. Neuropathologically, AD is defined by the extracellular accumulation of misfolded β-amyloid (Aβ) peptides into senile plaques and the intraneuronal formation of neurofibrillary tangles composed of hyperphosphorylated tau (p-tau). Aβ is generated through the sequential cleavage of amyloid precursor protein (APP) by β-secretase (BACE1) and γ-secretase, contributing to synaptic dysfunction and neuronal loss, alongside p-tau-driven tangle assembly, particularly in familial AD variants[1,3]. Despite decades of intensive investigation into these hallmark pathways, current pharmacological interventions offer only symptomatic relief or modest delays in cognitive decline; no treatments or medications have been shown to cure or stop AD pathology.
In recent years, research attention has shifted toward the non-neuronal milieu, where glial cells, including microglia, astrocytes, and oligodendrocytes, maintain central nervous system (CNS) homeostasis and support neuronal integrity through immune surveillance, synaptic modulation, blood-brain barrier (BBB) maintenance, and myelin preservation[4-6]. Dysregulation of glial cells has been shown to profoundly influence AD progression.
In AD, microglia initially confer neuroprotection by phagocytosing Aβ and clearing debris, yet chronic activation triggers a proinflammatory phenotype that amplifies Aβ and tau pathology and induces neurotoxic “A1” astrocytes[7]. Reactive astrocytes compromise BBB integrity and secrete cytokines that exacerbate neuroinflammation and synaptic loss. Emerging evidence also implicates oligodendrocyte dysfunction and altered progenitor cell dynamics in myelin disruption and metabolic insufficiency within AD brains. Mechanistically, these glial responses are orchestrated by key signaling cascades, such as the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), the NOD-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome, and the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) axis, offering novel molecular targets for therapeutic intervention[6,8].
In this review, we first outline the physiological functions of microglia, astrocytes, and oligodendrocytes in the healthy CNS. We then critically summarize their dynamic phenotypic shifts and associated regulatory mechanisms in AD pathogenesis. Finally, we discuss emerging glia-targeted strategies aimed at restoring neuron homeostasis, mitigating neuroinflammation, and preserving synaptic function, and we highlight translational challenges and future directions for glia-based therapies in AD.
GLIAL CELLS IN THE NERVOUS SYSTEM
Microglia
Microglia are the resident innate immune cells and specialized macrophages of the CNS, originating from primitive yolk sac macrophages during embryogenesis and persisting throughout life as vigilant sentinels[9,10]. Beyond immune surveillance, microglia engage in dynamic crosstalk with other CNS cell types to orchestrate critical developmental processes[11-13]. During neurogenesis, they guide the migration of inhibitory cortical interneurons, modulate axonal growth trajectories, and support the survival of layer V pyramidal neurons[14,15]. A defining function of microglia is activity-dependent synaptic refinement, whereby they selectively prune redundant synapses to optimize neural network efficiency[16,17]. In the mature brain, microglia maintain homeostasis by monitoring the neurovascular unit, regulating cerebral blood flow, and supporting metabolic demands[18,19]. Emerging evidence also implicates microglia in clearing apoptotic debris and dysfunctional synapses through interactions with astrocytes, oligodendrocytes, and vascular cells[20,21]. Their capacity to release both neurotrophic factors and proinflammatory mediators underscores their dual role in maintaining physiological function and mediating pathological responses in the CNS [Figure 1].
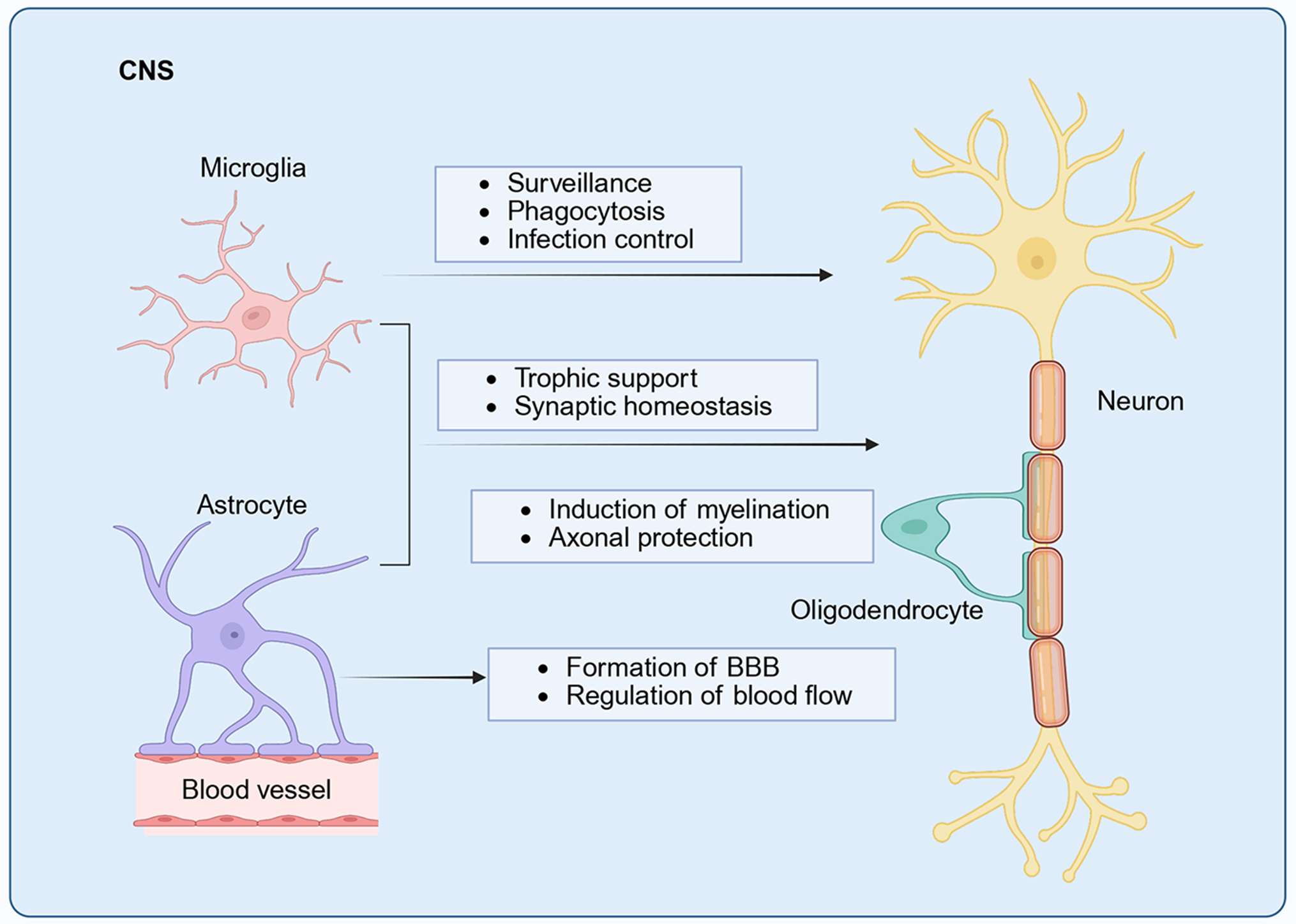
Figure 1. Functions of glial cells in the CNS. Under physiological conditions, microglia act as resident immune cells, engaging in immune surveillance, phagocytosis of cellular debris, and infection control. Astrocytes contribute to the formation of the BBB, regulate cerebral blood flow, and maintain synaptic homeostasis and neurotrophic support. Oligodendrocytes facilitate myelin formation and support the efficient propagation of bioelectrical signals through saltatory conduction, thereby safeguarding normal neuronal function. [Created in BioRender. 2, 1. (2025) https://BioRender.com/5gn4zwg]. CNS: Central nervous system; BBB: blood-brain barrier.
Astrocytes
Astrocytes are the most abundant glial population in the CNS and exhibit structural and functional diversity essential for neural circuit optimization. They are classified into protoplasmic and fibrous subtypes based on their localization in gray and white matter, respectively, and extend processes that contact synapses, blood vessels, and nodes of Ranvier[22]. Regional astrocyte subpopulations are further shaped by local microenvironmental cues and epigenetic programming[23-25]. Astrocytes far surpass their historical role as mere “glial glue”[26]. They actively regulate synaptogenesis through thrombospondin and SPARC secretion[27,28], modulate neurotransmitter homeostasis via glutamate and gamma-aminobutyric acid (GABA) recycling[29,30], and preserve BBB integrity through AQP4-enriched endfeet[31]. Under inflammatory conditions, astrocytes can also clear cellular debris and upregulate MHC-II expression[32,33], highlighting their neuroimmunological duality [Figure 1].
Oligodendrocytes
Oligodendrocytes and oligodendrocyte progenitor cells (OPCs) provide electrical insulation and metabolic support to CNS axons[34]. OPCs persist throughout life and differentiate into myelinating oligodendrocytes, forming lipid-rich sheaths that facilitate saltatory conduction[35,36]. Myelination exhibits activity-dependent plasticity, allowing adaptive refinement of neural circuits[37,38]. Beyond insulation, oligodendrocytes transfer lactate to axons and buffer extracellular potassium, thereby sustaining axonal integrity and preventing degeneration[39-41]. Through lifelong myelin remodeling and supporting mechanisms, oligodendrocytes maintain CNS resilience during development, adulthood, and nerve repair following injury[42] [Figure 1].
ALZHEIMER’S DISEASE PATHOLOGY
AD is driven by multiple neuropathological processes. Aβ is widely distributed in AD brains. Extracellular
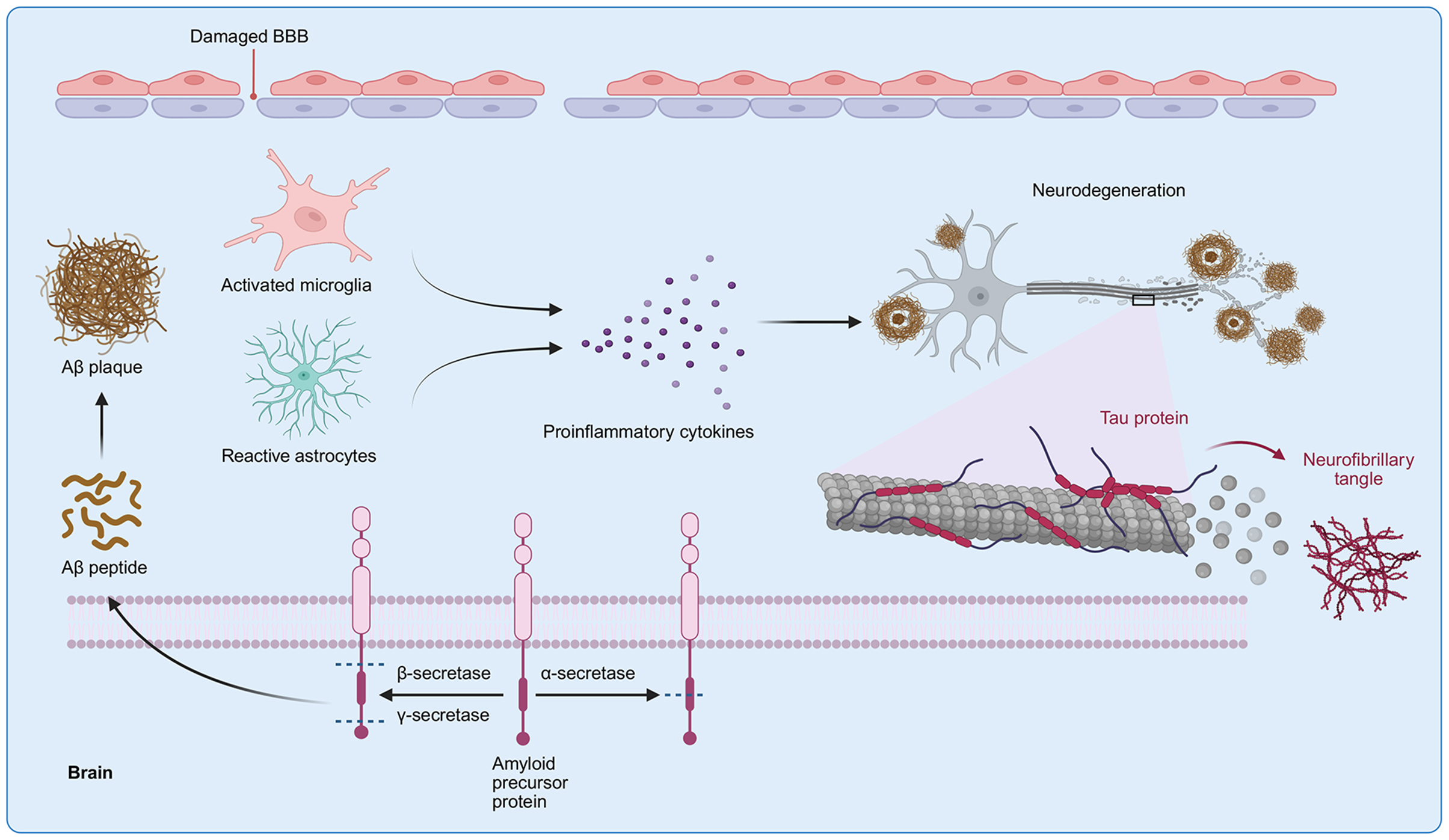
Figure 2. Pathological cascade of AD. Aberrant proteolytic cleavage of APP generates Aβ peptides with strong aggregation propensity. These peptides accumulate extracellularly to form insoluble Aβ plaques, which trigger microglial activation and astrocytic reactivity. Activated glial cells release proinflammatory cytokines, creating a neurotoxic microenvironment. Meanwhile, intracellular tau undergoes pathological hyperphosphorylation, leading to NFT formation. The synergy of Aβ-driven neuroinflammation, tau pathology, and glial activation ultimately disrupts BBB integrity and drives progressive neurodegeneration in the brain parenchyma. [Created in BioRender. Y, S. (2025) https://BioRender.com/4lv920b]. AD: Alzheimer’s disease; APP: amyloid precursor protein; Aβ: amyloid-β; NFT: neurofibrillary tangle; BBB: blood-brain barrier.
Synaptic dysfunction precedes neuronal loss in AD, with affected regions showing an estimated 30%-40% reduction in synaptic density[53,54]. The basal forebrain cholinergic system is particularly vulnerable, exhibiting decreased choline acetyltransferase activity and cortical acetylcholine depletion, both of which contribute to memory impairment[55,56]. Hippocampal atrophy observed on magnetic resonance imaging (MRI) reflects advanced neurodegeneration[57,58]. Concurrently, cerebral amyloid angiopathy (CAA), present in up to 95% of AD cases, deposits Aβ in leptomeningeal and cortical vessels, impairing cerebrovascular regulation and increasing the risk of hemorrhage[59,60].
Chronic neuroinflammation in AD is mediated by activated microglia and astrocytes responding to Aβ and tau pathology[61]. While initially protective, sustained release of proinflammatory cytokines (IL-1β, TNF-α, IL-6, IL-18) and complement factors creates a neurotoxic milieu that exacerbates neuronal damage[62-64]. Astrocytic upregulation of glial fibrillary acidic protein (GFAP) marks astrogliosis and has prognostic significance in disease progression[65,66]. Reactive microglia colocalize with amyloid plaques, underscoring their role in plaque-associated inflammation[67,68].
Aging remains the dominant risk factor for AD, with cellular senescence contributing to disease through the senescence-associated secretory phenotype (SASP), characterized by the release of proinflammatory and matrix-degrading molecules[69-71]. In AD, accumulation of senescent cells is accompanied by increased oxidative stress, mitochondrial dysfunction, and activation of inflammatory cascades[72,73], which in turn accelerate Aβ deposition and tau phosphorylation via a deleterious feedback loop. Additional drivers of senescence include impaired proteostasis, DNA damage, telomerase deficiency, and telomere shortening[74,75]. Collectively, AD pathogenesis reflects intricate interactions among protein misfolding, cellular stress responses, and impaired homeostatic mechanisms, highlighting the need for therapies that target these interconnected pathways.
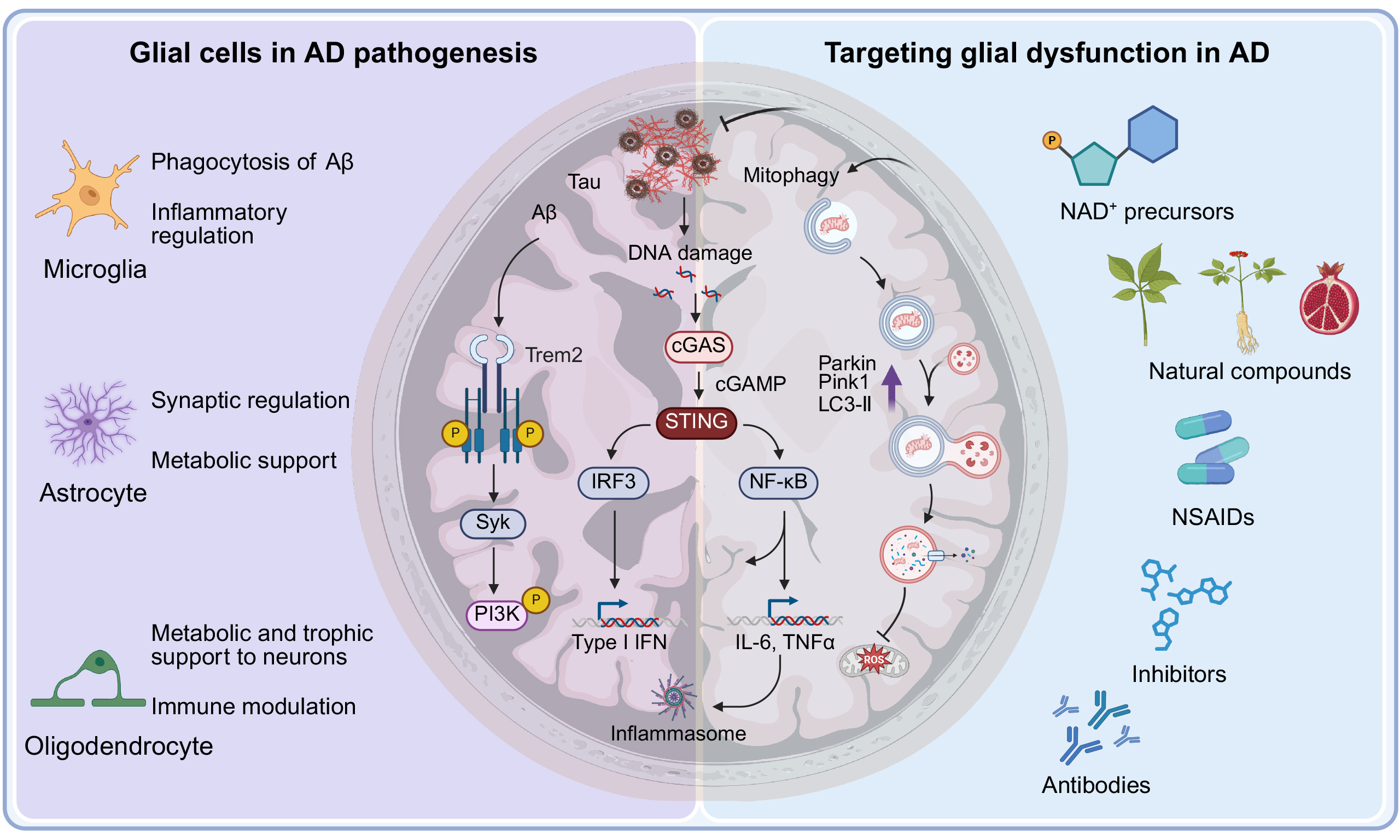
GLIAL CELLS IN AD PATHOGENESIS
Microglia in AD
Microglial proliferation is a hallmark of AD-related neuroinflammation and correlates with cognitive decline[76]. Increased microglial density, driven by upregulation of colony-stimulating factor 1 receptor (CSF1R), has been observed in APP/PS1, 3×TgAD, and 5×FAD mouse models, where it is associated with plaque load and behavioral deficits[77-82]. Pharmacological or genetic inhibition of CSF1R reduces microglial numbers, synaptic loss, and cognitive impairment without markedly altering Aβ burden[79].
Microglial phagocytosis modulates disease progression by engulfing Aβ plaques, synapses, and even entire neurons[83]. This process is orchestrated by receptors and risk genes including TREM2 and APOE[84]. APOE opsonizes Aβ to facilitate uptake[85], while TREM2 engagement promotes plaque compaction and restricts new seeding[85-88]. Early-stage phagocytosis may confer neuroprotection, but excessive or misdirected engulfment in later stages contributes to synaptic loss and neuronal death, underscoring the therapeutic complexity of modulating this function[89-91].
Under homeostatic conditions, microglia exhibit a ramified morphology and secrete minimal inflammatory mediators[92]. In AD, however, Aβ and p-tau activate and polarize microglia into diverse functional states. The classical M1 phenotype produces IL-1β, TNF-α, and IL-6, driving neuroinflammation and neuronal damage, whereas the M2 phenotype releases IL-4, IL-10, and TGF-β, supporting tissue repair and neuroprotection[93-96]. Recent single-cell and single-nucleus RNA sequencing (scRNA-seq and snRNA-seq) studies have revealed spatial and temporal heterogeneity beyond the M1/M2 paradigm, identifying disease-associated microglia (DAM) localized near Aβ plaques. DAM exhibit activated TREM2 signaling and enrichment of genes related to lipid metabolism and phagocytosis[97-100], indicating the therapeutic potential of targeting TREM2 or other DAM regulators. Microglia can thus transition from protective guardians to drivers of neurodegeneration[101].
In early AD, microglia clear Aβ via phagocytosis, a process enhanced by peripheral anti-Aβ antibodies that cross the blood-brain barrier and opsonize plaques[102]. However, chronic exposure to Aβ oligomers and phosphorylated tau drives a sustained proinflammatory state. Aβ binds TLR4, activating MyD88-dependent NF-κB signaling, while scavenger receptors (e.g., CD36) amplify cytokine release and oxidative burst[103]. Early TLR3 stimulation with poly(I:C) paradoxically suppresses this cycle, reducing plaques and inflammation in APP/PS1 mice[104]. Downstream, NF-κB upregulates TNF-α and IL-6, while apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC)-dependent NLRP3 inflammasome activation triggers caspase-1-mediated cleavage of pro-IL-1β into mature IL-1β[105,106]. These cytokines recruit astrocytes, which secrete additional inflammatory mediators, establishing a feed-forward loop of glial hyperactivation that undermines synaptic integrity and accelerates neuronal loss. Activated microglia also generate reactive oxygen species (ROS) and nitric oxide (NO), causing oxidative damage to lipids, proteins, and DNA, thereby contributing to synaptic dysfunction[107,108] and accelerating neuronal injury and loss[109,110]. Neuronal apoptosis is closely linked to this inflammatory milieu[111]. TNF-α engages neuronal death receptors to activate caspase-8 and downstream executioner caspases, while ROS/NO disrupt mitochondrial integrity, promote cytochrome c release, and activate intrinsic apoptotic pathways[107,112,113].
Microglial energy metabolism mirrors their activation state. Under physiological conditions, microglia predominantly rely on mitochondrial oxidative phosphorylation (OXPHOS) to meet energy needs and support surveillance functions[114]. Upon activation, they undergo metabolic reprogramming toward aerobic glycolysis, which provides rapid ATP and biosynthetic precursors for inflammation. In AD, this glycolytic shift becomes constitutive, persisting even in the absence of acute stimuli[115,116]. AD microglia exhibit elevated lactate production and acidification of the microenvironment, further impairing mitochondrial respiration[117,118]. Lipid droplet accumulation, commonly observed in AD microglia, also disrupts mitochondrial function and fosters a proinflammatory phenotype[119]. Several molecular mechanisms link mitochondrial dysfunction to altered microglial metabolism[120]. A key regulator is hypoxia-inducible factor 1-alpha (HIF-1α), stabilized under oxidative stress and promoting transcription of glycolytic enzymes[121]. Conversely, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a master regulator of mitochondrial biogenesis and function, is downregulated in APP/PS1 and 3×TgAD mouse models, correlating with impaired mitochondrial biogenesis and reduced respiratory capacity[122-124].
Cellular senescence, a state of irreversible cell-cycle arrest accompanied by the SASP, is increasingly implicated in AD[69,125]. In aged and APP/PS1 mouse brains, microglia exhibit DNA damage and telomere attrition caused by Aβ-induced oxidative stress and prolonged cytokine exposure[71]. These stressors activate the p53-p21 and p16INK4a-Rb pathways, driving microglial senescence[126]. Senescent microglia secrete SASP factors that perpetuate chronic neuroinflammation; elevated levels of these proteins have been detected in the brains, cerebrospinal fluid, and serum of AD patients[127-131]. Moreover, replicative senescence in Aβ-laden APP/PS1 mice exacerbates plaque deposition and synaptic deficits[132]. While senolytic approaches targeting p16INK4a-positive microglia have demonstrated efficacy in reducing neuroinflammation and improving cognition in other neurodegenerative models[70], a standardized set of microglial senescence markers remains undefined. Future studies should integrate longitudinal analyses of human brain tissue with single-cell profiling to establish robust senescence signatures and assess the therapeutic potential of senolytics in AD [Figure 3].
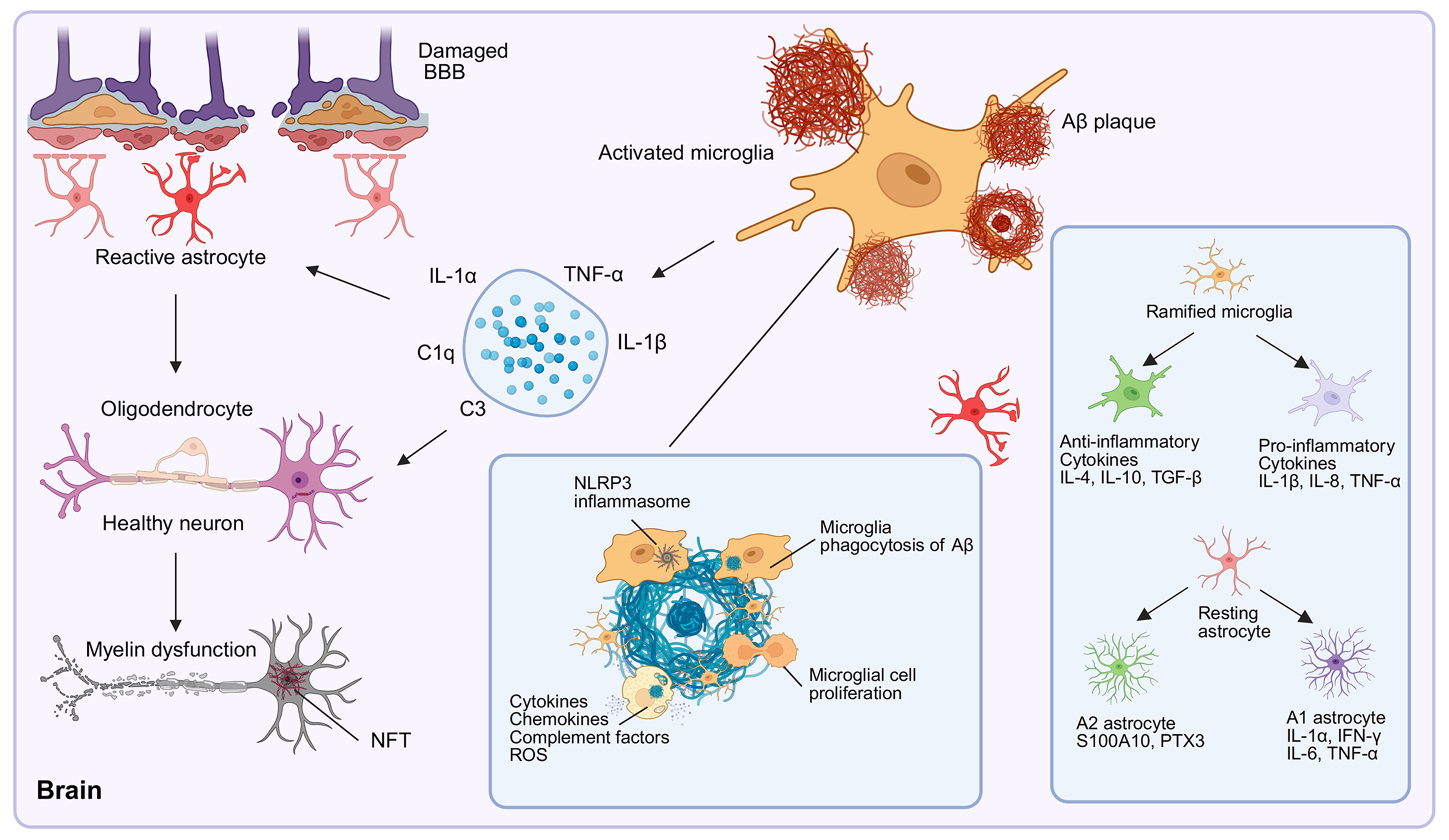
Figure 3. Role of glial cells in AD pathogenesis. Microglia transition from a ramified morphology to an activated state in an attempt to phagocytose Aβ. Chronic activation prompts microglia to release proinflammatory cytokines, ROS, and complement factors. This neuroinflammation promotes astrocytic reactivity, driving their polarization into proinflammatory A1 or potentially anti-inflammatory A2 subtypes, thereby contributing to astrogliosis. The interplay among activated glia, Aβ plaques, NFTs, BBB disruption, and inflammatory mediators (including those derived from the NLRP3 inflammasome) drives neuronal damage and disease progression, highlighting the dual neuroprotective and neurotoxic roles of glial cells in AD pathogenesis. [Created in BioRender. 1, 1. (2025) https://BioRender.com/w3zqhhf]. AD: Alzheimer’s disease; Aβ: amyloid-β; ROS: reactive oxygen species; NFT: neurofibrillary tangle; NFTs: neurofibrillary tangles; BBB: blood-brain barrier; NLR: NOD-like receptor; NLRP3: the NLR family pyrin domain containing 3.
Astrocytes in AD
Astrocytes are a heterogeneous glial population with diverse morphologies and functions that are essential for CNS homeostasis[133]. Under physiological conditions, they provide structural and metabolic support to neurons, regulate BBB integrity, modulate synaptic activity, recycle neurotransmitters, and maintain ion balance[133,134].
In AD, astrocytes undergo morphological, transcriptional, and functional remodeling, termed “reactive astrogliosis”, in response to Aβ and tau pathology[135]. Although reactive astrocytes were first described more than a century ago, their precise roles in CNS pathology, regeneration, and aging remain unresolved and continue to be debated. Reactive astrocytes are often categorized into two broad phenotypes: proinflammatory A1 and neuroprotective A2[136]. A1 astrocytes secrete neurotoxic factors (e.g., IL-1β,
In AD, reactive astrocytes exhibit hypertrophy of the soma and processes, increased expression of cytoskeletal proteins such as GFAP and vimentin, and retraction of fine processes[139]. Transcriptomic studies show upregulation of innate immune genes, including lipocalin 2 (LCN2) and serpin peptidase inhibitor alpha-1-antichymotrypsin (SERPINA3N), as well as enzymes in the cholesterol biosynthesis pathway. These alterations precede overt cognitive decline and correlate with amyloid plaque and neurofibrillary tangle burden[140,141].
Astrocytes also secrete proinflammatory mediators that amplify microglial activation and recruit peripheral immune cells[142,143]. Furthermore, they express complement proteins and pattern-recognition receptors (PRRs) such as TLR4, which facilitate Aβ recognition and inflammasome activation[144]. Crosstalk between astrocytes and microglia establishes a feed-forward loop: astrocyte-derived cytokines prime microglia for heightened inflammatory responses, while microglial factors reinforce astrocytic reactivity[145,146].
Astrocyte endfeet enwrap cerebral vessels, supporting tight junctions and BBB integrity through polarized aquaporin-4 (AQP4) localization and secretion of vascular‐stabilizing factors[147]. In AD, reactive astrocytes release matrix metalloproteinases (MMPs) and proinflammatory cytokines that degrade basement membrane components and disrupt tight junction proteins, thereby increasing BBB permeability and promoting leukocyte infiltration[7,148]. Loss of AQP4 polarization further impairs glymphatic clearance of Aβ and tau, facilitating their extracellular accumulation[149].
Perisynaptic astrocytes play a key role in neurotransmitter clearance and synaptic plasticity[150]. In AD, reactive astrocytes exhibit increased expression of monoamine oxidase-B (MAO-B), leading to elevated GABA synthesis and enhanced tonic inhibition, which contributes to cognitive impairment[151]. Additionally, astrocytic secretion of ceramides and other lipid mediators can directly damage synapses[152]. In contrast, A2-like astrocytes release neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF), and help maintain extracellular potassium homeostasis, thereby supporting neuronal viability[153-155]. The overall impact of astrocytes on synaptic health in AD reflects the dynamic balance between these opposing phenotypic states.
The widely used binary classification of reactive astrocytes into “A1” (neurotoxic) and “A2” (neuroprotective) subtypes has provided a useful framework for studying astrocyte heterogeneity. However, subsequent studies, including recent reviews, emphasize that this dichotomy oversimplifies the diverse and context-dependent phenotypes of reactive astrocytes, particularly in chronic neurodegenerative conditions such as AD[138,156]. For example, single-cell and single-nucleus RNA sequencing of mouse models and human AD brains has revealed a continuum of stage-dependent transcriptomic states that do not align neatly with the classical A1-A2 model. We therefore recommend that studies on reactive astrocytes integrate multiple molecular and functional readouts - ideally obtained in vivo - together with multivariate analyses and direct evaluation of their effects on disease-defining hallmarks in AD models [Figure 3].
Oligodendrocytes in AD
Oligodendrocytes, once considered passive bystanders in AD, are now recognized as active participants in neurodegeneration. scRNA-seq of the oligodendrocyte lineage in mice has revealed a developmental continuum from OPCs through immature to mature oligodendrocytes[157]. Both OPCs and mature oligodendrocytes interact closely with the neuroinflammatory milieu and Aβ-mediated toxicity, underscoring their active involvement in AD pathogenesis[158]. Before proliferating, OPCs must migrate toward lesion sites[159]. In AD, however, the OPC pool becomes progressively depleted. Their migration along blood-vessel scaffolds is impeded by extracellular matrix stiffening and amyloid-plaque barriers, leading OPCs to cluster rather than infiltrate lesions[160]. As a reservoir for new oligodendrocytes, OPCs are indispensable for myelin formation and repair[161]. In AD, Aβ peptides induce dysfunction and death of both OPCs and mature oligodendrocytes in vitro and in vivo[162]. Transcriptomic profiling of post-mortem dementia brains has revealed disrupted signaling cascades that drive progressive loss of OPC differentiation[163]. Furthermore, myelin debris shed by damaged oligodendrocytes accumulates extracellularly and strongly inhibits OPC differentiation[164]. The loss of myelin sheaths during demyelination compromises synaptic transmission and disrupts learning and memory[164,165]. Interestingly, a recent mouse study demonstrated concurrent myelin degeneration and repair, showing a robust loss of pre-existing myelin alongside increased oligodendrogenesis and remyelination during disease progression[166]. Mechanistically, myelin dysfunction exacerbates Aβ pathology by trapping β-secretase within axonal swellings, thereby enhancing cortical APP cleavage[167].
Emerging evidence identifies OPC senescence as a key driver of demyelination in AD. Post-mortem analysis revealed significantly higher numbers of senescent OPCs in the inferior parietal cortex of late-onset AD brains compared with MCI brains[70]. In an AD mouse model, Olig2- and NG2-expressing OPCs exhibited an aging-like phenotype, with elevated p21 and p16INK4a expression and increased senescence-associated β-galactosidase activity[70]. Senescent OPCs adopt a SASP rich in IL-6 and ROS, thereby amplifying neuroinflammation and secondary microglial activation[168,169]. Notably, in APP/PS1 mice, intermittent senolytic treatment (Dasatinib plus Quercetin) reduced plaque-associated senescent OPCs, attenuated neuroinflammation and Aβ burden, and improved hippocampus-dependent memory, supporting the therapeutic potential of targeting OPC senescence in AD[70].
In response to injury or amyloid and tau pathology, oligodendrocytes acquire a distinct disease-associated transcriptional signature marked by upregulation of immune-related genes (e.g., Serpina3n and C4b), suggesting engagement in protease inhibition and complement-mediated clearance[170-172]. Additional enriched pathways include TNF receptor signaling (Tnfrsf1a), interleukins (Il1β, Il33), heme oxygenase-1 (Hmox1), tumor necrosis factor (Tnf), and MHC class I molecules (H2-D1, H2-K1, B2m), indicating a shift toward immune surveillance and antigen presentation[172,173]. These oligodendrocyte states emerge in parallel with plaque deposition and may reflect a reactive adaptation to chronic injury and aging. Serpina3n encodes a serine protease inhibitor that can neutralize granzyme B, potentially protecting oligodendrocytes from CD8+ T cell-mediated cytotoxicity[170]. Similarly, C4b may facilitate cellular debris clearance but can also drive complement-dependent synaptic pruning and neurodegeneration[170].
Oligodendrocytes are crucial for neuronal health by providing metabolic support and ensuring efficient myelination. Reactive oligodendrocytes may relinquish these canonical roles, leading to impaired axonal conduction and neuronal energy deficits. As AD progresses, glial cells may undergo further functional changes, possibly shifting from protective states toward exhaustion or dysfunction. For instance, OPCs adjacent to Aβ plaques can undergo senescence and secrete proinflammatory mediators, worsening tissue damage[70]. Motif enrichment analysis of disease-associated oligodendrocyte transcripts has identified nuclear factor κB (NF-κB), signal transducer and activator of transcription (STAT), and interferon regulatory factor (IRF) family transcription factors as key regulators of this reactive state[171]. Notably, oligodendrocytes express high levels of amyloid processing genes - including APP, BACE1, PSEN1, and PSEN2 - in both mouse and human RNA-sequencing datasets[174,175]. Conditional deletion of BACE1 in oligodendrocytes reduces the amyloid plaque burden, underscoring their contribution to Aβ production[176,177]. However, oligodendrocyte states in human AD brains diverge from those in mouse models, emphasizing the need for species-specific mechanistic studies[100] [Figure 3].
MOLECULAR MECHANISMS AND SIGNALING PATHWAYS OF GLIAL CELLS IN AD
Glial cells in AD orchestrate a multilayered immune response that begins with the recognition of endogenous danger signals and culminates in chronic neuroinflammation, synaptic dysfunction, and neuronal loss. The following sections expand on each major axis - recognition, signaling hubs, effector release, pathological states, and modulatory receptors - highlighting mechanistic details, temporal dynamics, and therapeutic implications [Figure 4].
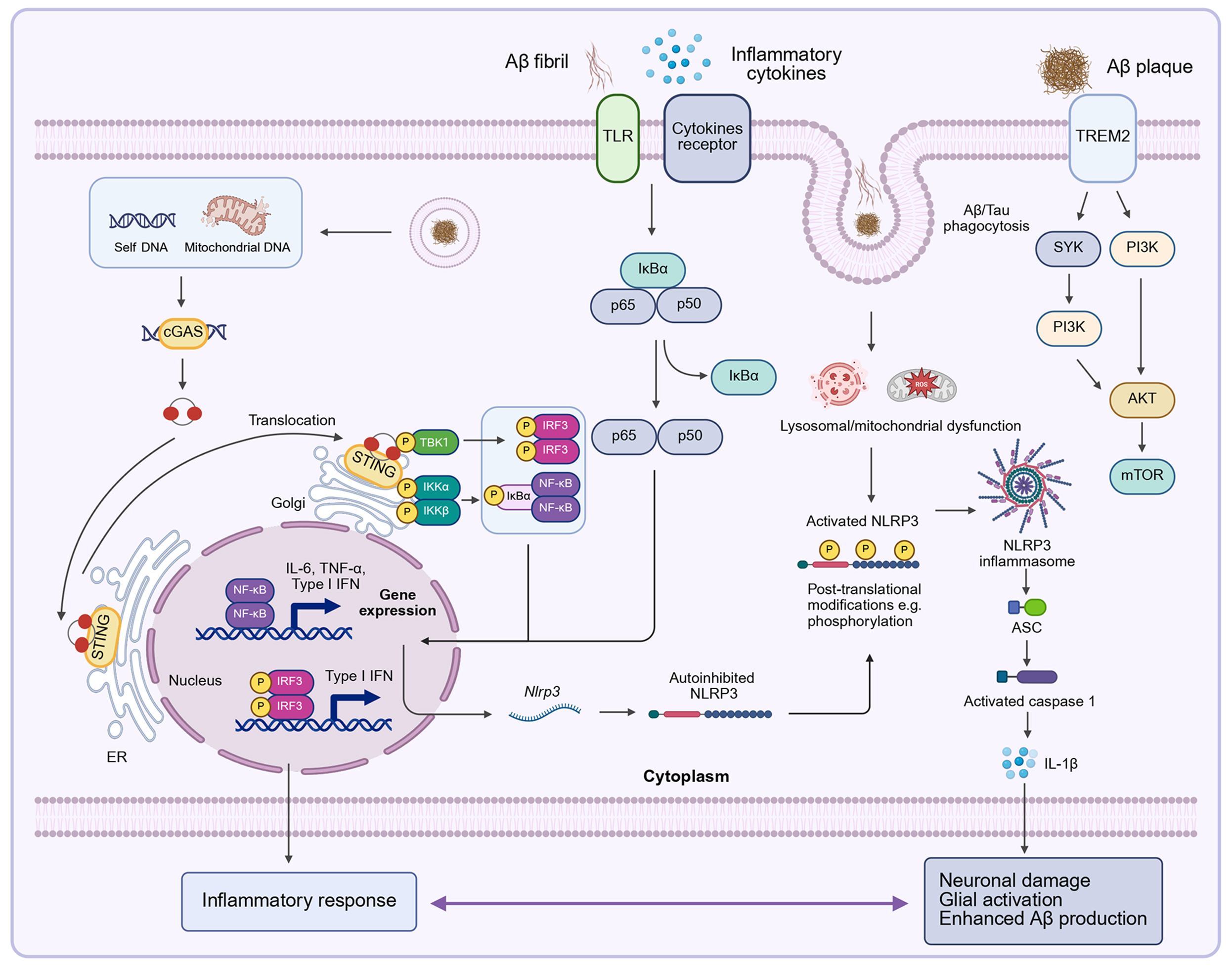
Figure 4. Molecular mechanisms and signaling pathways in AD. Multiple signaling pathways contribute to AD pathology, particularly those linked to neuroinflammation, neuronal death, and Aβ deposition. (1) cGAS-STING pathway: Cytosolic leakage of mtDNA or self-DNA activates cGAS, generating cGAMP that binds STING. This triggers TBK1-mediated IRF3 phosphorylation and NF-κB activation (via IκBα degradation and p65/p50 nuclear translocation), inducing type I interferons and proinflammatory cytokines (e.g., TNF-α, IL-6); (2) NLRP3 inflammasome pathway: DAMPs (e.g., Aβ fibrils) activate NLRP3, promoting ASC speck formation and caspase-1 cleavage. Activated caspase-1 converts pro-IL-1β into its mature form, amplifying neuroinflammation. This process is influenced by mitochondrial and lysosomal dysfunction and regulated by post-translational modifications; (3) TREM2 signaling: Aβ plaques engage TREM2 receptors on microglia, initiating SYK/PI3K/AKT phosphorylation cascades that enhance phagocytosis (Aβ/tau clearance) and support microglial survival. Dysregulated TREM2 impairs these functions, exacerbating plaque accumulation. Crosstalk among these pathways creates feed-forward loops that reinforce glial activation, neuronal damage, and further Aβ production. [Created in BioRender. 1, 1. (2025) https://BioRender.com/x4z0nw6]. AD: Alzheimer’s disease; Aβ: amyloid-β; cGAS: cyclic GMP-AMP synthase; STING: stimulator of interferon genes; mtDNA: mitochondrial DNA; cGAMP: 2’,3’-cyclic GMP-AMP; NF-κB: nuclear factor-κB; NLR: NOD-like receptor; NLRP3: NLR family pyrin domain containing 3.
Recognition of damage and DAMPs
Neurons and glia experiencing proteotoxic and metabolic stress release a spectrum of damage-associated molecular patterns (DAMPs), including oligomeric Aβ, misfolded tau, extracellular ATP, oxidized phospholipids, mitochondrial DNA (mtDNA), nuclear chromatin fragments, HMGB1, and S100 proteins[178,179]. Each DAMP engages specific PRRs on microglia and astrocytes.
Within minutes of exposure, Aβ oligomers induce the formation of the TLR2-TLR6-MyD88 complex, leading to NF-κB activation[178]. Simultaneously, fibrillar Aβ acts as an opsonin, providing a secondary activation signal for NLRP3 inflammasome assembly[64]. Extracellular tau species bind to TLR4, activating p38 and ERK1/2-MAPKs, which further amplify NF-κB signaling[179]. Microglial P2X7 receptors detect extracellular ATP released from dying cells or dysfunctional astrocytes, inducing K+ efflux, a potent trigger for NLRP3 inflammasome assembly[180]. In AD brains, cytosolic mtDNA and nuclear fragments accumulate due to impaired mitophagy and genotoxic stress. cGAS binds to this DNA, producing cGAMP, which activates STING and initiates its downstream signaling cascade[181]. Complement activation further amplifies danger signals: C1q and C3 are upregulated in the AD cortex and cluster around plaques and tangles, tagging synapses for microglial elimination[182,183]. The cleavage products C3a and C5a act as anaphylatoxins, and their receptors (C3aR and C5aR1) synergize with TLRs to enhance microglial inflammatory responses[184,185]. Experimental studies underscore the pathological role of complement. Genetic knockout of C3 in APP/PS1 mice preserves dendritic spines and reduces synaptic pruning[186], while pharmacological inhibition of C5aR1 (e.g., PMX205) decreases astrocytic GFAP expression and mitigates neuronal loss[187].
Core signaling pathways
Recognition signals converge on several central hubs, including NF-κB, cGAS-STING, the NLRP3 inflammasome, and JAK-STAT pathways, that coordinate the neuroinflammatory response in AD. NF-κB serves as a master regulator of proinflammatory gene expression. Downstream of MyD88, STING, or cytokine receptors, NF-κB is released from IκBα inhibition, translocates into the nucleus, and promotes transcription of numerous proinflammatory genes, including TNF-α, IL-6, pro-IL-1β, pro-IL-18, NLRP3, and iNOS[178]. Sustained NF-κB activity correlates with worsened cognitive performance in AD models.
The cGAS-STING pathway acts as a critical sensor of cytosolic DNA. Upon binding DNA, cGAS generates the second messenger cGAMP, which binds STING and triggers its translocation from the ER to the Golgi apparatus. Activated STING recruits TBK1, which phosphorylates the transcription factor IRF3, promoting its dimerization, nuclear translocation, and transcription of type I interferons (IFN-I), primarily IFN-β. Concurrently, STING activates the IKK complex, particularly IKKβ, resulting in NF-κB activation. The resulting IFN-β orchestrates an antiviral-like glial response by upregulating interferon-stimulated genes (ISGs) that modulate phagocytosis and antigen presentation[188]. While IFN-I signaling can initially enhance Aβ clearance, chronic activation contributes to synaptic dysfunction and memory deficits, indicating a narrow therapeutic window for targeting this pathway.
The NLRP3 inflammasome requires a two-step activation: priming by NF-κB and a secondary trigger such as K+ efflux, ROS, or lysosomal damage. Assembly of the NLRP3/ASC/caspase-1 complex initiates the autocatalytic activation of caspase-1. Active caspase-1 cleaves pro-IL-1β and pro-IL-18 into their mature, secreted forms (IL-1β and IL-18). Released ASC specks can further seed Aβ aggregation, perpetuating a vicious cycle of inflammation and proteopathy[64]. Pharmacological inhibition with compounds such as MCC950 has been shown to attenuate neuronal loss and cognitive decline in AD models.
The JAK-STAT pathway translates cytokine signaling into transcriptional programs. Activation of IFNAR phosphorylates STAT1/2, forming ISGF3 complexes that regulate hundreds of genes involved in antigen processing and phagocytosis. IL-6R-STAT3 signaling promotes acute-phase responses, while IL-10R-STAT3 drives glial cells toward suppression of proinflammatory programs[189]. Dysregulation of JAK-STAT signaling in AD contributes both to impaired resolution of inflammation and to inappropriate immunosuppression[190].
Effector mediators
Glial activation generates a complex cytokine and chemokine milieu that critically shapes the neuroinflammatory environment in AD. Among these mediators, proinflammatory cytokines - including TNF-α, IL-1β, IL-6, IL-18, and IFN-β - can reach nanomolar concentrations in AD brains[191]. These cytokines impair synaptic plasticity (by inhibiting LTP), disrupt BBB tight junctions, and recruit peripheral monocytes. In contrast, anti-inflammatory cytokines such as IL-4, IL-10, IL-13, and TGF-β are secreted by A2 astrocytes and regulatory T cells. Notably, IL-10 reduces microglial TNF-α production and enhances phagocytic clearance of Aβ, highlighting its therapeutic potential[191,192].
Chemokines also play crucial roles in leukocyte trafficking and microglial function. For instance, CCL2 (MCP-1) expression is elevated around amyloid plaques, recruiting CCR2+ monocytes that differentiate into CNS macrophages. The CX3CL1/CX3CR1 axis maintains homeostatic microglial motility and neuron-glia communication; its dysregulation correlates with excessive synaptic pruning[193].
Pathological cellular states
Chronic exposure to DAMPs and cytokines drives glia into maladaptive states characterized by oxidative stress, mitochondrial dysfunction, and defective autophagy. Aβ upregulates NOX2/4 and iNOS, generating ROS and RNS that oxidize mitochondrial proteins and lipids, impair electron transport, and trigger mtDNA release, thereby activating cGAS and NLRP3 signaling[120,194]. Impaired PINK1/Parkin-dependent mitophagy limits clearance of dysfunctional mitochondria, sustaining ROS production and inflammasome activation[194].
AD‐linked mutations in PSEN1/2 elevate lysosomal pH, blocking autophagosome-lysosome fusion and causing Aβ/tau accumulation[195,196]. GWAS variants in PICALM and CLU impair vesicle trafficking and enzyme activity, leading to enlarged endolysosomal compartments in glia[197,198]. Hyperactive mTORC1 (via phosphorylated S6K1) suppresses ULK1/BECN1, while TFEB downregulation reduces lysosomal biogenesis. Pharmacological TFEB activation restores lysosomal hydrolase activity and promotes clearance of intracellular Aβ and tau in cell models[199,200].
Modulatory receptors and phagocytic pathways
Glial responses are further modulated by receptors that balance clearance and inflammation. TREM2 binds APOE and Aβ, activating DAP12/SYK-PI3K/AKT signaling to support microglial survival, proliferation, and phagocytosis[201]. TREM2 R47H variants diminish these functions, leading to greater plaque burden and neuronal damage[90]. GWAS have identified TREM2 variants (particularly R47H) that increase late-onset Alzheimer’s disease (LOAD) risk by impairing microglial activity[202,203]. In early AD, TREM2 promotes microglial clustering around amyloid plaques and enhances Aβ clearance, whereas in later stages, TREM2 deficiency or mutation (e.g., R47H) compromises plaque compaction, leading to larger deposits and increased neuronal injury[204].
CD33/Siglec receptors recruit SHP phosphatases to intracellular ITIM motifs, attenuating Src kinase activation downstream of DAMP receptors. In AD cortex, CD33 overexpression is associated with reduced microglial Aβ clearance[205]. Scavenger receptors (CD36, SR-A1) mediate uptake of oxidized lipids and Aβ fibrils[206]. However, chronic stimulation promotes lipid droplet accumulation in microglia, skewing them toward a proinflammatory, glycolytic phenotype.
TARGETING GLIAL CELLS TO TREAT AD
NAD+ precursors
NAD+ is an essential coenzyme in redox reactions, regulating mitochondrial energy metabolism and DNA repair. Its levels decline with age and in neurodegenerative diseases, including AD. The NAD+ precursor nicotinamide riboside (NR) is converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT), after which NMN is converted to NAD+ by nicotinamide mononucleotide adenylyltransferase (NMNAT)[207,208]. Recent studies have highlighted the therapeutic potential of NR and NMN in ameliorating AD pathology[69,209].
Supplementation with NR has been shown to restore NAD+ levels, reduce neuroinflammation and DNA damage through NAD+-dependent activation of sirtuins (e.g., SIRT3, SIRT6), and thereby decrease tau phosphorylation and improve memory in AD mouse models[75]. Mitochondrial dysfunction, a hallmark of AD, is associated with impaired energy production, oxidative damage, and neuronal apoptosis[210]. NMN administration enhances mitochondrial biogenesis and activity through NAD+-dependent activation of sirtuins, leading to reduced tau phosphorylation in human neuronal cells and improved memory in tauopathy models, including transgenic nematodes and mice[194]. Furthermore, NR promotes mitophagy through the PINK1-Parkin pathway, suppresses the cGAS-STING pathway, reduces cellular senescence and SASP, and thereby decreases neuroinflammation and improves motor function in neurodegeneration-prone AD and Atm-/- mice[71,211] [Table 1].
Therapeutic approaches and compounds in AD treatment
| Therapeutic approach | Agent/compound | Mechanism of action | References |
| NAD+ precursors | NR, NMN | Elevate NAD+ levels; reduce oxidative stress; restore hippocampal synaptic plasticity; decrease tau phosphorylation; enhance mitochondrial function; improve cognitive performance | [75,194,211] |
| Natural compounds & metabolites | Curcumin | Inhibits NF-κB and MAPK signaling, attenuating neuroinflammation; acts as a potent antioxidant, reducing lipid/protein oxidation; inhibits Aβ and tau aggregation | [214,215] |
| Resveratrol | Activates SIRT1 and suppresses NF-κB signaling; downregulates MMP-9; inhibits Aβ aggregation; promotes mitochondrial biogenesis | [217,218] | |
| Quercetin | Blocks NF-κB/MAPK signaling, reducing proinflammatory cytokines; enhances microglial phagocytosis of Aβ; reduces oxidative stress | [219-221] | |
| Melatonin | Suppresses IL-1β-mediated glial activation; upregulates Aβ-degrading enzymes; enhances BDNF/CREB-dependent synaptic plasticity; protects mitochondria integrity | [222,224,225] | |
| Urolithin A (UA) | Scavenges ROS; promotes mitochondrial quality control (mitophagy); reduces Aβ aggregation; enhances lysosomal function; improves learning and memory | [194,229,230] | |
| NSAIDs | Ibuprofen | Inhibits COX activity, reducing prostaglandin levels; suppresses microglial activation; reduces Aβ plaque load; improves cognitive performance in APP models | [237,238] |
| AchE inhibitors | Donepezil | Inhibits AChE, increasing synaptic acetylcholine, which improves cognition and ADL scores | [243,245] |
| Rivastigmine | Inhibits both AChE and BuChE, increasing acetylcholine; improves cognition and daily function | [246,247] | |
| Galantamine | Inhibits AChE and allosterically modulates α7-nAChR, increasing acetylcholine; slows cognitive and functional decline | [244,248] | |
| NLRP3 inflammasome inhibitors | MCC950 | Directly blocks NLRP3, inhibiting IL-1β/IL-18 release; reduces microgliosis and synaptic loss | [256] |
| cGAS-STING inhibitors | H-151 | Covalently antagonizes STING, suppressing type I IFN and NF-κB signaling; attenuates neuroinflammation and Aβ pathology | [258] |
| Immunotherapeutic strategies | CAD106 | Aβ1-6 peptide conjugated to a Qβ carrier; induces anti-Aβ antibodies, associated with reduced amyloid burden on PET | [264,265] |
| AADvac1 | Tau-derived peptide vaccine that elicits anti-tau antibodies, reducing tau aggregation and improving cognition | [266,267] | |
| Aducanumab | Human IgG1 monoclonal antibody targeting aggregated Aβ N-terminus; reduces amyloid plaque burden | [268,269] | |
| Lecanemab | Human monoclonal antibody against soluble Aβ protofibrils; reduces plaque burden and slows cognitive decline | [268,272] | |
| AL002c | Agonistic anti-TREM2 monoclonal antibody enhances microglial phagocytosis of Aβ; improves synaptic markers | [277,279] |
Natural compounds and metabolites
Mounting evidence supports the pivotal role of chronic neuroinflammation in AD progression. This recognition has driven increased interest in plant-derived natural compounds and microbiome-derived metabolites as potential modulators of glial activation and neuroinflammatory responses[212-215] [Table 1].
Resveratrol, a polyphenol abundant in red grapes, peanuts, and various berries, has been shown to reduce cerebrospinal fluid (CSF) levels of matrix metalloproteinase-9 (MMP9) while exerting potent anti-inflammatory effects. Mechanistically, resveratrol activates SIRT1, inhibits NF-κB signaling, and enhances mitochondrial function. Collectively, these actions contribute to its neuroprotective effects, promote adaptive immune activation, and modestly improve cognition in AD patients[216-218].
Quercetin, a flavonoid present in apples, onions, and many vegetables, also demonstrates potent anti-inflammatory activity. It inhibits microglial NF-κB and MAPK signaling, thereby reducing the production of proinflammatory cytokines such as IL-1β and TNF-α[219]. In addition, quercetin enhances microglial phagocytosis and promotes neuronal survival by reducing oxidative stress and preserving mitochondrial integrity[220,221].
Melatonin, a pineal-derived indoleamine, has also been implicated in AD therapy[222]. In rodent models, melatonin administration suppresses IL-1β-induced astrocyte and microglial activation, decreases proinflammatory cytokine expression, and improves learning and memory performance[223]. Accumulating evidence indicates that melatonin enhances Aβ clearance by upregulating neprilysin and insulin-degrading enzyme, while also promoting synaptic plasticity via BDNF and CREB signaling pathways[224,225]. Furthermore, melatonin stabilizes mitochondrial membrane potential, reduces cytochrome c release, and prevents neuronal apoptosis, underscoring its multifaceted neuroprotective potential in AD[226].
Urolithin A (UA), a gut microbiota-derived metabolite of ellagitannins, has recently emerged as a promising therapeutic candidate in AD models[227,228]. Owing to its phenolic hydroxyl groups, UA exhibits strong antioxidant properties that neutralize ROS and reduce oxidative stress. Beyond its antioxidant activity, UA exerts immunomodulatory effects by enhancing microglial phagocytosis and reducing neuroinflammation in APP/PS1 mice[194,229]. Mechanistically, UA promotes mitophagy through the PINK1/Parkin pathway and induces the expression of lysosomal histone proteases, particularly CTSZ, which is frequently disrupted in AD brains. Long-term UA treatment has been shown to improve learning, memory, and olfactory function in transgenic AD mice while concurrently reducing Aβ deposition and tau pathology[230]. Additionally, UA activates SIRT1 and inhibits mTOR, thereby enhancing neuronal resilience to aging-related stressors, including D-galactose-induced senescence[231]. In 3×TgAD mice, UA not only reduced amyloid burden but also significantly enhanced spatial memory performance. In vitro, UA stimulates mitophagy and promotes Aβ clearance in neuronal cultures. Notably, in normal aging wild-type mice, UA supplementation has been associated with lifespan extension and improved mitochondrial function[232].
Nonsteroidal anti-inflammatory drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been investigated for their neuroprotective potential in AD due to their ability to inhibit cyclooxygenase (COX) enzymes and thus reduce prostaglandin-mediated neuroinflammation[233]. Prostaglandins, produced by COX-1 and COX-2, contribute to glial activation, cytokine release, and BBB permeability[234,235]. In APP-overexpressing mouse models, chronic ibuprofen treatment reduces Aβ plaque deposition and attenuates microglial activation[236-238]. Similarly, other NSAIDs, such as indomethacin and celecoxib, decrease γ-secretase activity and lower brain Aβ₄₂ levels in the Tg2576 mouse model[239]. Early small-scale clinical studies suggested that indomethacin might slow cognitive decline in mild-to-moderate AD[240]. However, subsequent large, double-blind, randomized, placebo-controlled trials of nonselective NSAIDs (e.g., naproxen, diclofenac) and COX-2 selective inhibitors (e.g., celecoxib, rofecoxib) failed to demonstrate significant cognitive benefits in AD patients[241]. These disappointing outcomes may reflect the importance of timing: anti-inflammatory interventions may be more effective if initiated during preclinical or prodromal stages of AD, before extensive pathology has developed[242] [Table 1].
Acetylcholinesterase inhibitors
Cholinergic hypofunction led to the development of acetylcholinesterase inhibitors (AChEIs) to increase synaptic acetylcholine levels. Donepezil, rivastigmine, and galantamine are FDA-approved AChEIs that modestly improve cognition, activities of daily living, and global clinical status in mild-to-moderate AD[243,244]. Donepezil (5-10 mg/day) inhibits acetylcholinesterase (AChE), elevating cortical acetylcholine and producing small but consistent improvements on the ADAS-Cog and ADCS-ADL scales over 12-24 weeks; higher doses (23 mg/day) do not confer additional efficacy and are associated with more adverse events[243,245]. Rivastigmine (6-12 mg/day), which inhibits both AChE and butyrylcholinesterase, improves cognitive and functional measures in international placebo-controlled trials[246,247]. Galantamine
NMDA receptor antagonists
Overactivation of N-methyl-D-aspartate (NMDA) receptors contributes to excitotoxicity and glial activation in AD[250]. Memantine, an uncompetitive NMDA receptor antagonist, reduces pathological calcium influx, thereby protecting neurons and modulating microglial and astrocytic responses[251]. In moderate-to-severe AD, memantine (10-20 mg/day) produces modest improvements in cognitive, functional, and behavioral outcomes compared with placebo, as demonstrated in meta-analyses of randomized trials[252,253]. By normalizing glutamatergic neurotransmission and reducing excitotoxic drive, memantine complements cholinergic therapies and represents one of the few currently licensed adjunctive agents for AD management[254] [Table 1].
NLRP3 inflammasome inhibitors
Preclinical studies demonstrate that pharmacological inhibition of NLRP3 reduces Aβ deposition, attenuates glial activation, and preserves cognitive function in animal models of AD[255]. For example, the small-molecule inhibitor MCC950 blocks NLRP3 assembly and has shown efficacy in both streptozotocin-induced and transgenic AD mouse models by lowering IL-1β levels, reducing microgliosis, and improving memory performance[256]. These findings support NLRP3 as a promising therapeutic target for disrupting the cycle of inflammation and neurodegeneration in AD [Table 1].
cGAS-STING antagonists
Inhibitors of the cGAS-STING pathway show significant therapeutic potential in neurodegenerative disorders. Pharmacological blockade with brain-penetrant cGAS inhibitors or the covalent STING antagonist H-151 suppresses IFN-I and NF-κB signaling, decreases expression of Tnfa, C1q, C3, and Il1a, and preserves synaptic integrity in both amyloid and tauopathy models[257,258]. Mechanistically, H-151 covalently targets Cys91 on STING, preventing dimerization and subsequent TBK1-IRF3 signaling[259], while novel cGAS inhibitors (e.g., acetaldehyde derivatives) prevent cGAS-DNA binding and phase separation[260]. Notably, mTORC2-mediated phosphorylation of cGAS at Ser37 regulates its chromatin localization, suggesting that epigenetic modulation may represent an alternative strategy[261]. Refining selective cGAS-STING inhibitors with favorable CNS pharmacokinetics, particularly enhancing BBB penetration and minimizing off-target effects, remains critical for clinical translation. Future directions include developing dual inhibitors that block both cGAS enzymatic activity and STING activation, as well as exploring combination therapies with immunomodulatory agents [Table 1].
Immunotherapeutic strategies
Glial cells play both pathogenic and protective roles in AD pathology, driving the development of active and passive immunotherapies targeting Aβ, tau, or immune receptors on microglia[262,263] [Table 1].
Active immunization employs antigenic peptides to stimulate endogenous antibody production. CAD106, which displays multiple copies of Aβ1-6 on a bacteriophage Qβ carrier, demonstrated safety and a correlation between antibody tiers and PET imaging in a 90-week Phase 2 trial in patients with mild AD, with reduced amyloid load on PET scans[264,265]. AADvac1 targets pathological tau epitopes, and its ongoing Phase 2 trial is evaluating effects on cognitive measures and tau biomarkers in mild-to-moderate AD[266,267].
Passive immunization involves the administration of monoclonal antibodies that directly clear toxic proteins. Aducanumab, approved by the FDA in 2021, selectively binds aggregated Aβ and reduces plaque burden[268,269]. In Phase 3 trials, EMERGE showed a significant reduction in clinical decline, whereas ENGAGE did not replicate this effect[270,271]. In January 2024, the manufacturer announced the global discontinuation of aducanumab development and commercialization. Lecanemab, a monoclonal antibody targeting soluble Aβ protofibrils, received accelerated approval in 2023 after demonstrating both plaque reduction and slower cognitive decline[272]. Across multiple clinical trials, lecanemab was generally well tolerated[272-274]; the most common adverse events were infusion-related reactions, amyloid-related imaging abnormalities (ARIA), and headache. ARIA is subdivided into ARIA-E (edema) and ARIA-H (hemosiderin deposition, including microhemorrhage and superficial siderosis), both of which are associated with cerebral amyloid angiopathy (CAA) and ApoE ε4, and may also occur spontaneously in AD[275]. ARIA-E was more frequent in the lecanemab group than in placebo, but was generally mild to moderate on imaging and typically occurred within the first 3-6 months of treatment[276]. Awareness of incidence, monitoring, and management strategies is therefore critical for clinicians, patients, and caregivers to ensure optimal patient care.
Antibodies against receptors such as TREM2 have also shown promise in preclinical models by enhancing microglial phagocytosis, reducing Aβ plaques, and improving synaptic markers[201,277]. Anti-TREM2 antibodies such as AL002c are designed to enhance microglial clearance of Aβ but face unique translational challenges[277]. Chronic TREM2 agonism may drive microglia toward a neurotoxic DAM stage, as observed in mouse models. Highaffinity AL002c mimetics have been reported to increase peri-plaque neuritic tau pathology and neuritic dystrophy without reducing Aβ plaque burden, and to exacerbate synapse loss in an amyloidosis mouse model where AD-tau was injected to induce Aβ-dependent tau seeding and spreading[278,279]. These findings highlight the context- and disease stage-dependent nature of microglial function: administration that is too early (when microglia remain largely quiescent) or too late (when they are already proinflammatory) may tip the balance toward detrimental rather than beneficial effects. It is also important to note that not all TREM2 agonist antibodies are equivalent, and whether similar outcomes would be observed with other antibodies remains uncertain. By modulating glial immune functions and directly clearing pathogenic aggregates, these immunotherapeutic modalities offer complementary strategies to attenuate neuroinflammation and slow disease progression in AD.
CONCLUSION
Glial cells, including microglia, astrocytes, and oligodendrocytes, have emerged as central regulators of AD pathogenesis, wielding both neuroprotective and neurotoxic influences that influence disease onset and progression[5,280]. Microglia orchestrate innate immune responses: in early stages, they clear Aβ and secrete neurotrophic factors, but chronic activation promotes the release of IL-1β, TNF-α, and reactive oxygen species, exacerbating synaptic loss and neuronal death[4,281]. Astrocytes support BBB integrity, neurotransmitter recycling, and metabolic function; however, the “A1” reactive subtype contributes to neuroinflammation and impairs Aβ clearance[138,280]. Oligodendrocytes, once overlooked, can adopt disease-associated states that modulate immune signaling and myelin homeostasis, thereby affecting neuronal network stability[157].
Therapeutic strategies must reflect the complexity of glial biology, moving beyond amyloid- and tau-centric monotherapies toward integrated combination regimens. These regimens should incorporate inflammasome and innate immune modulators, such as small-molecule inhibitors targeting NLRP3 or cGAS-STING pathways, which have demonstrated preclinical efficacy in reducing glial inflammation and preserving cognition[282,283]. Metabolic and senescence‐targeted agents, including NAD+ precursors like NR or NMN, combined with senolytics, also hold promise for restoring glial energy metabolism, reducing SASP burden, and restoring phagocytic capacity[71,194]. Additionally, immune‐modulatory antibodies and vaccines, encompassing both passive and active immunotherapies directed at Aβ, tau, or glial receptors such as TREM2 and CD33, offer opportunities to recalibrate microglial states toward enhanced debris clearance and repair[283,284].
Precision medicine approaches may further optimize therapy by stratifying patients based on glial biomarkers, such as CSF or plasma cytokine panels, PET imaging of TSPO or MAO-B, and single-cell transcriptomic profiles of microglial and astrocyte states[139,285]. Longitudinal monitoring of glial activation with emerging PET ligands may also help determine treatment timing and dosage.
Next-generation experimental models are critical for translating these insights. Patient-derived iPSC systems, ranging from simple glial monocultures to complex 3D neural tissues, reproduce AD-related Aβ/tau pathology and glial activation while linking cellular phenotypes to donor cognitive traits, despite inherent heterogeneity[139,286]. Human-mouse chimeric brains, in which iPSC-derived microglia colonize immunodeficient rodents, replicate human-specific glial responses to Aβ and reveal time-dependent disease-associated states[287,288]. Future hybrid platforms - such as organ-on-chip systems incorporating perfused microvessels and adaptive immune cells - combined with high-resolution single-cell and spatial omics, will enable precise mapping of glial state dynamics and temporally controlled perturbations[289]. Such models will help identify intercellular nodes whose modulation can shift glial communities from pathogenic to protective states.
Nevertheless, significant challenges remain. Robust glial biomarkers that predict treatment response, improved in vitro and in vivo models that better recapitulate human glial diversity, and delivery platforms (e.g., nanoparticles, viral vectors) with high glial specificity are urgently needed. Moreover, the interplay between central and peripheral immunity in AD remains underexplored; modulating systemic myeloid cells may synergize with CNS-focused glial therapies.
In summary, a glia-centric paradigm for AD - one that acknowledges the dual roles of microglia, astrocytes, and oligodendrocytes - offers the potential for durable disease modification. Continued investment in glial biology, coupled with advances in biomarker development and drug delivery, may transform AD from an inexorable decline into a manageable condition with preserved cognition and quality of life.
DECLARATIONS
Acknowledgments
The authors thank all other lab members for valuable discussions. All figures were created using BioRender.com with its images and schematic components, and the corresponding copyright licenses have been obtained. The graphic abstract was also created in BioRender. 1, 1. (2025) https://BioRender.com/hxoiejh.
Authors’ contributions
Writing of the original draft: Zhu Q, Song Y, Qian Y
Revised and edited the manuscript: Hou Y, Xue J, Zhu Q, Song Y, Qian Y, Zhang R
All authors reviewed and approved the final version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This study was supported by the National Key R&D Program of China (2022YFA1103203 to Xue J), the National Natural Science Foundation of China (Grants 82171405, 82471614 to Hou Y and 32270639 to Xue J), the Natural Science Foundation of Shanghai (23ZR1465600 to Hou Y and 23ZR1468200 to Xue J), the Non-profit Central Research Institute Fund of the Chinese Academy of Medical Sciences (2024-JKCS-19 to Hou Y and Xue J), the Fundamental Research Funds for the Central Universities (22120250374 to Hou Y and Xue J), the Lingang Laboratory (LG-QS-202205-10 to Hou Y), and Peak Disciplines (Type IV) of Institutions of Higher Learning in Shanghai.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Sasaguri H, Hashimoto S, Watamura N, et al. Recent advances in the modeling of Alzheimer’s disease. Front Neurosci. 2022;16:807473.
2. Götz J, Bodea LG, Goedert M. Rodent models for Alzheimer disease. Nat Rev Neurosci. 2018;19:583-98.
3. Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270-81.
5. Kedia S, Simons M. Oligodendrocytes in Alzheimer’s disease pathophysiology. Nat Neurosci. 2025;28:446-56.
6. Uddin MS, Lim LW. Glial cells in Alzheimer’s disease: from neuropathological changes to therapeutic implications. Ageing Res Rev. 2022;78:101622.
7. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9:42.
8. Sadick JS, O’Dea MR, Hasel P, Dykstra T, Faustin A, Liddelow SA. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron. 2022;110:1788-1805.e10.
9. Bennett ML, Bennett FC. The influence of environment and origin on brain resident macrophages and implications for therapy. Nat Neurosci. 2020;23:157-66.
10. Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol. 2021;39:251-77.
11. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314-8.
12. Thion MS, Ginhoux F, Garel S. Microglia and early brain development: an intimate journey. Science. 2018;362:185-9.
13. Borst K, Dumas AA, Prinz M. Microglia: immune and non-immune functions. Immunity. 2021;54:2194-208.
14. Ueno M, Fujita Y, Tanaka T, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16:543-51.
15. Cserép C, Pósfai B, Dénes Á. Shaping neuronal fate: functional heterogeneity of direct microglia-neuron interactions. Neuron. 2021;109:222-40.
16. Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456-8.
17. Filipello F, Morini R, Corradini I, et al. The microglial innate immune receptor trem2 is required for synapse elimination and normal brain connectivity. Immunity. 2018;48:979-991.e8.
18. Bisht K, Okojie KA, Sharma K, et al. Capillary-associated microglia regulate vascular structure and function through PANX1-P2RY12 coupling in mice. Nat Commun. 2021;12:5289.
19. Császár E, Lénárt N, Cserép C, et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. J Exp Med. 2022:219.
20. Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269-73.
21. McAlpine CS, Park J, Griciuc A, et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature. 2021;595:701-6.
22. Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science. 2018;362:181-5.
23. Bayraktar OA, Bartels T, Holmqvist S, et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat Neurosci. 2020;23:500-9.
24. Wheeler MA, Clark IC, Tjon EC, et al. MAFG-driven astrocytes promote CNS inflammation. Nature. 2020;578:593-9.
25. Endo F, Kasai A, Soto JS, et al. Molecular basis of astrocyte diversity and morphology across the CNS in health and disease. Science. 2022;378:eadc9020.
27. Christopherson KS, Ullian EM, Stokes CC, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421-33.
28. Tsai HH, Li H, Fuentealba LC, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337:358-62.
29. Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227-38.
30. Koh W, Kwak H, Cheong E, Lee CJ. GABA tone regulation and its cognitive functions in the brain. Nat Rev Neurosci. 2023;24:523-39.
31. Díaz-Castro B, Robel S, Mishra A. Astrocyte endfeet in brain function and pathology: open questions. Annu Rev Neurosci. 2023;46:101-21.
32. Damisah EC, Hill RA, Rai A, et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv. 2020;6:eaba3239.
33. Sanmarco LM, Polonio CM, Wheeler MA, Quintana FJ. Functional immune cell-astrocyte interactions. J Exp Med. 2021:218.
34. Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871-927.
36. Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779-82.
37. Xin W, Chan JR. Myelin plasticity: sculpting circuits in learning and memory. Nat Rev Neurosci. 2020;21:682-94.
38. Xin W, Kaneko M, Roth RH, et al. Oligodendrocytes and myelin limit neuronal plasticity in visual cortex. Nature. 2024;633:856-63.
39. Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. 2002;36:855-68.
40. Fünfschilling U, Supplie LM, Mahad D, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517-21.
41. Looser ZJ, Faik Z, Ravotto L, et al. Oligodendrocyte-axon metabolic coupling is mediated by extracellular K+ and maintains axonal health. Nat Neurosci. 2024;27:433-48.
42. Stadelmann C, Timmler S, Barrantes-Freer A, Simons M. Myelin in the central nervous system: structure, function, and pathology. Physiol Rev. 2019;99:1381-431.
43. Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885-90.
44. Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 1994;13:45-53.
46. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791-800.
47. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913-7.
48. Bancher C, Brunner C, Lassmann H, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989;477:90-9.
49. Aragão Gomes L, Uytterhoeven V, Lopez-Sanmartin D, et al. Maturation of neuronal AD-tau pathology involves site-specific phosphorylation of cytoplasmic and synaptic tau preceding conformational change and fibril formation. Acta Neuropathol. 2021;141:173-92.
50. Ohm TG, Müller H, Braak H, Bohl J. Close-meshed prevalence rates of different stages as a tool to uncover the rate of Alzheimer’s disease-related neurofibrillary changes. Neuroscience. 1995;64:209-17.
51. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960-9.
52. Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271-88.
53. Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM. Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci. 2017;40:347-57.
54. Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 1991;81:428-33.
55. Sakurada T, Alufuzoff I, Winblad B, Nordberg A. Substance P-like immunoreactivity, choline acetyltransferase activity and cholinergic muscarinic receptors in Alzheimer’s disease and multi-infarct dementia. Brain Res. 1990;521:329-32.
56. Boyd WD, Graham-White J, Blackwood G, Glen I, McQueen J. Clinical effects of choline in Alzheimer senile dementia. Lancet. 1977;2:711.
57. Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol. 2012;11:170-8.
58. Ossenkoppele R, Pijnenburg YA, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain. 2015;138:2732-49.
59. Vidoni ED, Yeh HW, Morris JK, et al. Cerebral β-amyloid angiopathy is associated with earlier dementia onset in Alzheimer’s disease. Neurodegener Dis. 2016;16:218-24.
60. Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16:30-42.
61. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388-405.
62. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463-77.
63. Corbo RM, Businaro R, Scarabino D. Leukocyte telomere length and plasma interleukin-1β and interleukin-18 levels in mild cognitive impairment and Alzheimer’s disease: new biomarkers for diagnosis and disease progression? Neural Regen Res. 2021;16:1397-8.
64. Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674-8.
65. Chatterjee P, Pedrini S, Stoops E, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl Psychiatry. 2021;11:27.
66. Verberk IMW, Laarhuis MB, van den Bosch KA, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic-based cohort study. Lancet Healthy Longev. 2021;2:e87-95.
67. Hayes A, Thaker U, Iwatsubo T, Pickering-Brown SM, Mann DM. Pathological relationships between microglial cell activity and tau and amyloid beta protein in patients with Alzheimer’s disease. Neurosci Lett. 2002;331:171-4.
68. Dani M, Wood M, Mizoguchi R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain. 2018;141:2740-54.
69. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565-81.
70. Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22:719-28.
71. Hou Y, Wei Y, Lautrup S, et al. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci U S A. 2021:118.
72. de la Cueva M, Antequera D, Ordoñez-Gutierrez L, et al. Amyloid-β impairs mitochondrial dynamics and autophagy in Alzheimer’s disease experimental models. Sci Rep. 2022;12:10092.
73. Li R, Li Y, Zuo H, Pei G, Huang S, Hou Y. Alzheimer’s amyloid-β accelerates cell senescence and suppresses SIRT1 in human neural stem cells. Biomolecules. 2024;14:189.
74. Jaskelioff M, Muller FL, Paik JH, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102-6.
75. Hou Y, Lautrup S, Cordonnier S, et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A. 2018;115:E1876-85.
76. Malpetti M, Kievit RA, Passamonti L, et al. Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain. 2020;143:1588-602.
77. Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, McGeer PL. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res. 1994;639:171-4.
78. Gómez-Nicola D, Fransen NL, Suzzi S, Perry VH. Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci. 2013;33:2481-93.
79. Olmos-Alonso A, Schetters ST, Sri S, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain. 2016;139:891-907.
80. Sosna J, Philipp S, Albay R 3rd, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener. 2018;13:11.
81. Spangenberg EE, Lee RJ, Najafi AR, et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain. 2016;139:1265-81.
82. Dagher NN, Najafi AR, Kayala KM, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12:139.
83. Lepiarz-Raba I, Gbadamosi I, Florea R, Paolicelli RC, Jawaid A. Metabolic regulation of microglial phagocytosis: implications for Alzheimer’s disease therapeutics. Transl Neurodegener. 2023;12:48.
84. Andrews SJ, Renton AE, Fulton-Howard B, Podlesny-Drabiniok A, Marcora E, Goate AM. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. EBioMedicine. 2023;90:104511.
85. Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566-581.e9.
86. Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276-1290.e17.
87. Grubman A, Choo XY, Chew G, et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun. 2021;12:3015.
88. Parhizkar S, Arzberger T, Brendel M, et al. Author Correction: Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2024;27:1858-9.
89. Dejanovic B, Wu T, Tsai MC, et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat Aging. 2022;2:837-50.
90. Popescu AS, Butler CA, Allendorf DH, et al. Alzheimer’s disease-associated R47H TREM2 increases, but wild-type TREM2 decreases, microglial phagocytosis of synaptosomes and neuronal loss. Glia. 2023;71:974-90.
91. Gratuze M, Leyns CE, Sauerbeck AD, et al. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J Clin Invest. 2020;130:4954-68.
92. Cai Y, Liu J, Wang B, Sun M, Yang H. Microglia in the neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front Immunol. 2022;13:856376.
93. Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39:3-18.
94. Wu C, Yang L, Feng S, et al. Therapeutic non-invasive brain treatments in Alzheimer’s disease: recent advances and challenges. Inflamm Regen. 2022;42:31.
95. Guillot-Sestier MV, Doty KR, Town T. Innate immunity fights Alzheimer’s disease. Trends Neurosci. 2015;38:674-81.
96. Wang Q, Yao H, Liu W, et al. Microglia polarization in Alzheimer’s disease: mechanisms and a potential therapeutic target. Front Aging Neurosci. 2021;13:772717.
97. Jin W, Pei J, Roy JR, et al. Comprehensive review on single-cell RNA sequencing: a new frontier in Alzheimer’s disease research. Ageing Res Rev. 2024;100:102454.
98. van Olst L, Simonton B, Edwards AJ, et al. Microglial mechanisms drive amyloid-β clearance in immunized patients with Alzheimer’s disease. Nat Med. 2025;31:1604-16.
99. Martins-Ferreira R, Calafell-Segura J, Leal B, et al. The Human Microglia Atlas (HuMicA) unravels changes in disease-associated microglia subsets across neurodegenerative conditions. Nat Commun. 2025;16:739.
100. Zhou Y, Song WM, Andhey PS, et al. Author Correction: Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med. 2020;26:981.
101. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441-68.
102. Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916-9.
103. Momtazmanesh S, Perry G, Rezaei N. Toll-like receptors in Alzheimer’s disease. J Neuroimmunol. 2020;348:577362.
104. Wang S, Zhu T, Ni W, et al. Early activation of Toll-like receptor-3 reduces the pathological progression of Alzheimer’s disease in APP/PS1 mouse. Alzheimers Res Ther. 2023;15:33.
105. Cantero-Fortiz Y, Boada M. The role of inflammation in neurological disorders: a brief overview of multiple sclerosis, Alzheimer’s, and Parkinson’s disease’. Front Neurol. 2024;15:1439125.
106. Gurel B, Cansev M, Sevinc C, et al. Early Stage Alterations in CA1 extracellular region proteins indicate dysregulation of IL6 and iron homeostasis in the 5XFAD Alzheimer’s disease mouse model. J Alzheimers Dis. 2018;61:1399-410.
107. Xu S, Yang P, Qian K, et al. Modulating autophagic flux via ROS-responsive targeted micelles to restore neuronal proteostasis in Alzheimer’s disease. Bioact Mater. 2022;11:300-16.
108. Aminzadeh M, Roghani M, Sarfallah A, Riazi GH. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int Immunopharmacol. 2018;54:78-85.
109. Deecke L, Homann J, Goldeck D, et al. No increase of CD8+ TEMRA cells in the blood of healthy adults at high genetic risk of Alzheimer’s disease. Alzheimers Dement. 2024;20:3116-8.
110. Huang R, Zhang Z, Shi Z, Yang Y, Sun J, Gao F. Ratiometric fluorescence imaging of lysosomal NO in living cells and mice brains with Alzheimer’s disease. Chem Commun. 2024;60:6793-6.
111. Saaoud F, Liu L, Xu K, et al. Alzheimer’s disease as an auto-innate immune pathology with potential cell trans-differentiation and enhanced trained immunity in 3xTg-AD mouse model. J Alzheimers Dis. 2025;105:550-72.
112. Singh A, Tiwari S, Singh S. Pirh2 modulates the mitochondrial function and cytochrome c-mediated neuronal death during Alzheimer’s disease. Cell Death Dis. 2024;15:331.
113. Kumari S, Dhapola R, Reddy DH. Apoptosis in Alzheimer’s disease: insight into the signaling pathways and therapeutic avenues. Apoptosis. 2023;28:943-57.
114. Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal. 2014;20:353-71.
115. Yin F, Sancheti H, Patil I, Cadenas E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic Biol Med. 2016;100:108-22.
116. Baik SH, Kang S, Lee W, et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 2019;30:493-507.e6.
117. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015;6:6176.
118. McCommis KS, Finck BN. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J. 2015;466:443-54.
119. Zyśk M, Beretta C, Naia L, et al. Amyloid-β accumulation in human astrocytes induces mitochondrial disruption and changed energy metabolism. J Neuroinflammation. 2023;20:43.
120. Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15:30.
121. Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582:359-64.
122. Panes JD, Wendt A, Ramirez-Molina O, Castro PA, Fuentealba J. Deciphering the role of PGC-1α in neurological disorders: from mitochondrial dysfunction to synaptic failure. Neural Regen Res. 2022;17:237-45.
123. Jia WW, Lin HW, Yang MG, et al. Electroacupuncture activates AMPKα1 to improve learning and memory in the APP/PS1 mouse model of early Alzheimer’s disease by regulating hippocampal mitochondrial dynamics. J Integr Med. 2024;22:588-99.
124. Singulani MP, Pereira CPM, Ferreira AFF, et al. Impairment of PGC-1α-mediated mitochondrial biogenesis precedes mitochondrial dysfunction and Alzheimer’s pathology in the 3xTg mouse model of Alzheimer’s disease. Exp Gerontol. 2020;133:110882.
125. Bellelli F, Angioni D, Arosio B, Vellas B, De Souto Barreto P. Hallmarks of aging and Alzheimer’s disease pathogenesis: paving the route for new therapeutic targets. Ageing Res Rev. 2025;106:102699.
126. Gutta G, Mehta J, Kingston R, et al. DNA damage and senescence in the aging and Alzheimer’s disease cortex are not uniformly distributed. Biomedicines. 2024;12:1327.
127. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586.
128. Herdy JR, Traxler L, Agarwal RK, et al. Increased post-mitotic senescence in aged human neurons is a pathological feature of Alzheimer’s disease. Cell Stem Cell. 2022;29:1637-1652.e6.
129. Guerrero A, De Strooper B, Arancibia-Cárcamo IL. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021;44:714-27.
130. Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 2006;29:506-10.
131. Blum-Degen D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202:17-20.
132. Hu Y, Fryatt GL, Ghorbani M, et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 2021;35:109228.
133. Escartin C, Galea E, Lakatos A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312-25.
134. Jiwaji Z, Tiwari SS, Avilés-Reyes RX, et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat Commun. 2022;13:135.
135. Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia. 2018;66:637-53.
136. Fan YY, Huo J. A1/A2 astrocytes in central nervous system injuries and diseases: angels or devils? Neurochem Int. 2021;148:105080.
137. Kaur D, Sharma V, Deshmukh R. Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology. 2019;27:663-77.
138. Habib N, McCabe C, Medina S, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23:701-6.
139. Heneka MT, van der Flier WM, Jessen F, et al. Neuroinflammation in Alzheimer disease. Nat Rev Immunol. 2025;25:321-52.
140. Siddiqui T, Cosacak MI, Popova S, et al. Nerve growth factor receptor (Ngfr) induces neurogenic plasticity by suppressing reactive astroglial Lcn2/Slc22a17 signaling in Alzheimer’s disease. NPJ Regen Med. 2023;8:33.
141. Han X, Lei Q, Liu H, Zhang T, Gou X. SerpinA3N regulates the secretory phenotype of mouse senescent astrocytes contributing to neurodegeneration. J Gerontol A Biol Sci Med Sci. 2024;79:glad278.
142. Dhapola R, Hota SS, Sarma P, Bhattacharyya A, Medhi B, Reddy DH. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology. 2021;29:1669-81.
143. Giovannoni F, Quintana FJ. The role of astrocytes in CNS inflammation. Trends Immunol. 2020;41:805-19.
144. Verkhratsky A, Rodrigues JJ, Pivoriunas A, Zorec R, Semyanov A. Astroglial atrophy in Alzheimer’s disease. Pflugers Arch. 2019;471:1247-61.
145. Ju YH, Bhalla M, Hyeon SJ, et al. Astrocytic urea cycle detoxifies Aβ-derived ammonia while impairing memory in Alzheimer’s disease. Cell Metab. 2022;34:1104-1120.e8.
146. Chun H, Im H, Kang YJ, et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2- production. Nat Neurosci. 2020;23:1555-66.
147. Gao Y, Ye S, Tang Y, Tong W, Sun S. Brain cholesterol homeostasis and its association with neurodegenerative diseases. Neurochem Int. 2023;171:105635.
148. Zhao M, Jiang XF, Zhang HQ, et al. Interactions between glial cells and the blood-brain barrier and their role in Alzheimer’s disease. Ageing Res Rev. 2021;72:101483.
149. Feng S, Wu C, Zou P, et al. High-intensity interval training ameliorates Alzheimer’s disease-like pathology by regulating astrocyte phenotype-associated AQP4 polarization. Theranostics. 2023;13:3434-50.
150. Andersen JV, Schousboe A, Verkhratsky A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022;217:102331.
151. Alam J, Blackburn K, Patrick D. Neflamapimod: clinical phase 2b-ready oral small molecule inhibitor of p38α to reverse synaptic dysfunction in early Alzheimer’s disease. J Prev Alzheimers Dis. 2017;4:273-8.
152. Mi Y, Qi G, Vitali F, et al. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat Metab. 2023;5:445-65.
153. Zhu H, Xiao F, Xiao Y, et al. Targeting CB2R in astrocytes for Parkinson’s disease therapy: unraveling the Foxg1-mediated neuroprotective mechanism through autophagy-mediated NLRP3 degradation. J Neuroinflammation. 2023;20:304.
154. Glantz LA, Gilmore JH, Lieberman JA, Jarskog LF. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr Res. 2006;81:47-63.
155. Al-Ghraiybah NF, Wang J, Alkhalifa AE, et al. Glial cell-mediated neuroinflammation in Alzheimer’s disease. Int J Mol Sci. 2022;23:10572.
156. Grubman A, Chew G, Ouyang JF, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci. 2019;22:2087-97.
157. Brown TI, Carr VA, LaRocque KF, et al. Prospective representation of navigational goals in the human hippocampus. Science. 2016;352:1323-6.
158. Lau V, Ramer L, Tremblay MÈ. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat Commun. 2023;14:1670.
159. Skaper SD. Oligodendrocyte precursor cells as a therapeutic target for demyelinating diseases. Prog Brain Res. 2019;245:119-44.
160. Behrendt G, Baer K, Buffo A, et al. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia. 2013;61:273-86.
161. Gautier HO, Evans KA, Volbracht K, et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun. 2015;6:8518.
162. Lee JT, Xu J, Lee JM, et al. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123-31.
163. Pietrzak M, Papp A, Curtis A, et al. Gene expression profiling of brain samples from patients with Lewy body dementia. Biochem Biophys Res Commun. 2016;479:875-80.
164. Papuć E, Rejdak K. The role of myelin damage in Alzheimer’s disease pathology. Arch Med Sci. 2020;16:345-51.
165. Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6:22.
166. Chen JF, Liu K, Hu B, et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron. 2021;109:2292-2307.e5.
167. Depp C, Sun T, Sasmita AO, et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature. 2023;618:349-57.
168. Liu Y, Aguzzi A. NG2 glia are required for maintaining microglia homeostatic state. Glia. 2020;68:345-55.
169. Zhang SZ, Wang QQ, Yang QQ, et al. NG2 glia regulate brain innate immunity via TGF-β2/TGFBR2 axis. BMC Med. 2019;17:204.
170. Haile Y, Carmine-Simmen K, Olechowski C, Kerr B, Bleackley RC, Giuliani F. Granzyme B-inhibitor serpina3n induces neuroprotection in vitro and in vivo. J Neuroinflammation. 2015;12:157.
171. Kenigsbuch M, Bost P, Halevi S, et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat Neurosci. 2022;25:876-86.
172. Falcão AM, van Bruggen D, Marques S, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018;24:1837-44.
173. Kirby L, Jin J, Cardona JG, et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun. 2019;10:3887.
174. Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929-47.
175. Zhang Y, Sloan SA, Clarke LE, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37-53.
176. Rajani RM, Ellingford R, Hellmuth M, et al. Selective suppression of oligodendrocyte-derived amyloid beta rescues neuronal dysfunction in Alzheimer’s disease. PLoS Biol. 2024;22:e3002727.
177. Sasmita AO, Depp C, Nazarenko T, et al. Oligodendrocytes produce amyloid-β and contribute to plaque formation alongside neurons in Alzheimer’s disease model mice. Nat Neurosci. 2024;27:1668-74.
178. Venegas C, Heneka MT. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J. 2019;33:13075-84.
179. Pampuscenko K, Morkuniene R, Krasauskas L, Smirnovas V, Brown GC, Borutaite V. Extracellular tau stimulates phagocytosis of living neurons by activated microglia via Toll-like 4 receptor-NLRP3 inflammasome-caspase-1 signalling axis. Sci Rep. 2023;13:10813.
180. Yue N, Huang H, Zhu X, et al. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J Neuroinflammation. 2017;14:102.
181. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786-91.
182. Wu T, Dejanovic B, Gandham VD, et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 2019;28:2111-2123.e6.
183. Dejanovic B, Huntley MA, De Mazière A, et al. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron. 2018;100:1322-1336.e7.
184. Fonseca MI, Chu SH, Berci AM, et al. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J Neuroinflammation. 2011;8:4.
185. Gomez-Arboledas A, Carvalho K, Balderrama-Gutierrez G, et al. C5aR1 antagonism alters microglial polarization and mitigates disease progression in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun. 2022;10:116.
186. Shi Q, Chowdhury S, Ma R, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017:9.
187. Fonseca MI, Ager RR, Chu SH, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol. 2009;183:1375-83.
188. Ishikawa H, Barber GN. The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell Mol Life Sci. 2011;68:1157-65.
189. Sarapultsev A, Gusev E, Komelkova M, Utepova I, Luo S, Hu D. JAK-STAT signaling in inflammation and stress-related diseases: implications for therapeutic interventions. Mol Biomed. 2023;4:40.
190. Rusek M, Smith J, El-Khatib K, Aikins K, Czuczwar SJ, Pluta R. The role of the JAK/STAT signaling pathway in the pathogenesis of Alzheimer’s disease: new potential treatment target. Int J Mol Sci. 2023;24:864.
191. McAlpine FE, Lee JK, Harms AS, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34:163-77.
192. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481-7.
193. Wojcieszak J, Kuczyńska K, Zawilska JB. Role of chemokines in the development and progression of Alzheimer’s disease. J Mol Neurosci. 2022;72:1929-51.
194. Fang EF, Hou Y, Palikaras K, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401-12.
195. Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146-58.
196. Wang B, Martini-Stoica H, Qi C, et al. TFEB-vacuolar ATPase signaling regulates lysosomal function and microglial activation in tauopathy. Nat Neurosci. 2024;27:48-62.
197. Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088-93.
198. Moreau K, Fleming A, Imarisio S, et al. PICALM modulates autophagy activity and tau accumulation. Nat Commun. 2014;5:4998.
199. Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429-33.
200. Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926-37.
201. Ulrich JD, Ulland TK, Colonna M, Holtzman DM. Elucidating the role of TREM2 in Alzheimer’s Disease. Neuron. 2017;94:237-48.
202. Guerreiro R, Wojtas A, Bras J, et al; Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117-27.
203. Schmid CD, Sautkulis LN, Danielson PE, et al. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83:1309-20.
204. Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med. 2018;215:745-60.
205. Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631-43.
206. Xu S, Chaudhary O, Rodríguez-Morales P, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity. 2021;54:1561-1577.e7.
207. Yoshino J, Baur JA, Imai SI. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27:513-28.
208. Fang EF, Lautrup S, Hou Y, et al. NAD+ in aging: molecular mechanisms and translational implications. Trends Mol Med. 2017;23:899-916.
209. Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD+ metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021;22:119-41.
210. Picca A, Faitg J, Auwerx J, Ferrucci L, D’Amico D. Mitophagy in human health, ageing and disease. Nat Metab. 2023;5:2047-61.
211. Yang B, Dan X, Hou Y, et al. NAD+ supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell. 2021;20:e13329.
212. Shen Y, Liu F, Zhang M. Therapeutic potential of plant-derived natural compounds in Alzheimer’s disease: targeting microglia-mediated neuroinflammation. Biomed Pharmacother. 2024;178:117235.
213. Dey A, Bhattacharya R, Mukherjee A, Pandey DK. Natural products against Alzheimer’s disease: pharmaco-therapeutics and biotechnological interventions. Biotechnol Adv. 2017;35:178-216.
214. Shao S, Ye X, Su W, Wang Y. Curcumin alleviates Alzheimer’s disease by inhibiting inflammatory response, oxidative stress and activating the AMPK pathway. J Chem Neuroanat. 2023;134:102363.
215. Feng Q, Zhang X, Zhao X, et al. Intranasal delivery of pure nanodrug loaded liposomes for Alzheimer’s disease treatment by efficiently regulating microglial polarization. Small. 2024;20:e2405781.
216. Nguyen C, Coudeyre E, Boutron I, et al. Oral resveratrol in adults with knee osteoarthritis: a randomized placebo-controlled trial (ARTHROL). PLoS Med. 2024;21:e1004440.
217. Moussa C, Hebron M, Huang X, et al. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflammation. 2017;14:1.
218. Li C, Wang N, Zheng G, Yang L. Oral administration of resveratrol-selenium-peptide nanocomposites alleviates Alzheimer’s disease-like pathogenesis by inhibiting Aβ aggregation and regulating gut microbiota. ACS Appl Mater Interfaces. 2021;13:46406-20.
219. Zu G, Sun K, Li L, Zu X, Han T, Huang H. Mechanism of quercetin therapeutic targets for Alzheimer disease and type 2 diabetes mellitus. Sci Rep. 2021;11:22959.
220. Wang DM, Li SQ, Wu WL, Zhu XY, Wang Y, Yuan HY. Effects of long-term treatment with quercetin on cognition and mitochondrial function in a mouse model of Alzheimer’s disease. Neurochem Res. 2014;39:1533-43.
221. Khan H, Ullah H, Aschner M, Cheang WS, Akkol EK. Neuroprotective effects of quercetin in Alzheimer’s disease. Biomolecules. 2019;10:59.
222. Chen C, Yang C, Wang J, et al. Melatonin ameliorates cognitive deficits through improving mitophagy in a mouse model of Alzheimer’s disease. J Pineal Res. 2021;71:e12774.
223. Shen Y, Zhang G, Liu L, Xu S. Suppressive effects of melatonin on amyloid-beta-induced glial activation in rat hippocampus. Arch Med Res. 2007;38:284-90.
224. Zhang Z, Xue P, Bendlin BB, et al. Melatonin: a potential nighttime guardian against Alzheimer’s. Mol Psychiatry. 2025;30:237-50.
225. Li Y, Zhang J, Wan J, Liu A, Sun J. Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed Pharmacother. 2020;132:110887.
226. Ribeiro RFN, Santos MR, Aquino M, de Almeida LP, Cavadas C, Silva MMC. The therapeutic potential of melatonin and its novel synthetic analogs in circadian rhythm sleep disorders, inflammation-associated pathologies, and neurodegenerative diseases. Med Res Rev. 2025;45:1515-39.
227. D’Amico D, Andreux PA, Valdés P, Singh A, Rinsch C, Auwerx J. Impact of the natural compound urolithin A on health, disease, and aging. Trends Mol Med. 2021;27:687-99.
228. Kuerec AH, Lim XK, Khoo AL, et al. Targeting aging with urolithin A in humans: a systematic review. Ageing Res Rev. 2024;100:102406.
229. Gong Z, Huang J, Xu B, et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J Neuroinflammation. 2019;16:62.
230. Hou Y, Chu X, Park JH, et al. Urolithin A improves Alzheimer’s disease cognition and restores mitophagy and lysosomal functions. Alzheimers Dement. 2024;20:4212-33.
231. Chen P, Chen F, Lei J, Li Q, Zhou B. Activation of the miR-34a-mediated SIRT1/mTOR signaling pathway by urolithin A attenuates D-galactose-induced brain aging in mice. Neurotherapeutics. 2019;16:1269-82.
232. Ballesteros-Álvarez J, Nguyen W, Sivapatham R, Rane A, Andersen JK. Urolithin A reduces amyloid-beta load and improves cognitive deficits uncorrelated with plaque burden in a mouse model of Alzheimer’s disease. Geroscience. 2023;45:1095-113.
233. Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflammation. 2022;19:206.
234. Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC; Cache County Study Investigators. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the cache county study. Neurology. 2002;59:880-6.
235. Jordan F, Quinn TJ, McGuinness B, et al. Aspirin and other non-steroidal anti-inflammatory drugs for the prevention of dementia. Cochrane Database Syst Rev. 2020;4:CD011459.
236. Yan Q, Zhang J, Liu H, et al. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J Neurosci. 2003;23:7504-9.
238. Fielder E, Tweedy C, Wilson C, et al. Anti-inflammatory treatment rescues memory deficits during aging in nfkb1-/- mice. Aging Cell. 2020;19:e13188.
239. in t’ Veld BA, Ruitenberg A, Hofman A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515-21.
240. Rogers J, Kirby LC, Hempelman SR, et al. Clinical trial of indomethacin in Alzheimer’s disease. Neurology. 1993;43:1609-11.
241. Soininen H, West C, Robbins J, Niculescu L. Long-term efficacy and safety of celecoxib in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;23:8-21.
242. Sastre M, Gentleman SM. NSAIDs: how they work and their prospects as therapeutics in Alzheimer’s disease. Front Aging Neurosci. 2010;2:20.
243. Birks JS, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2018;6:CD001190.
244. Lim AWY, Schneider L, Loy C. Galantamine for dementia due to Alzheimer’s disease and mild cognitive impairment. Cochrane Database Syst Rev. 2024;11:CD001747.
245. Koh SH, Kwon HS, Choi SH, et al. Efficacy and safety of GV1001 in patients with moderate-to-severe Alzheimer’s disease already receiving donepezil: a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. Alzheimers Res Ther. 2021;13:66.
246. Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318:633-8.
247. Bastiat G, Plourde F, Motulsky A, et al. Tyrosine-based rivastigmine-loaded organogels in the treatment of Alzheimer’s disease. Biomaterials. 2010;31:6031-8.
248. Burns A, Bernabei R, Bullock R, et al. Safety and efficacy of galantamine (Reminyl) in severe Alzheimer’s disease (the SERAD study): a randomised, placebo-controlled, double-blind trial. Lancet Neurol. 2009;8:39-47.
249. Patwardhan AG, Belemkar S. An update on Alzheimer’s disease: immunotherapeutic agents, stem cell therapy and gene editing. Life Sci. 2021;282:119790.
250. Companys-Alemany J, Turcu AL, Schneider M, et al. NMDA receptor antagonists reduce amyloid-β deposition by modulating calpain-1 signaling and autophagy, rescuing cognitive impairment in 5XFAD mice. Cell Mol Life Sci. 2022;79:408.
251. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312-39.
252. McShane R, Westby MJ, Roberts E, et al. Memantine for dementia. Cochrane Database Syst Rev. 2019;3:CD003154.
253. Greenamyre JT, Maragos WF, Albin RL, Penney JB, Young AB. Glutamate transmission and toxicity in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12:421-30.
254. Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and management of dementia: review. JAMA. 2019;322:1589-99.
255. de Oliveira P, Cella C, Locker N, et al. Improved sleep, memory, and cellular pathological features of tauopathy, including the NLRP3 inflammasome, after chronic administration of trazodone in rTg4510 mice. J Neurosci. 2022;42:3494-509.
256. He XF, Xu JH, Li G, et al. NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2020;11:849.
257. Udeochu JC, Amin S, Huang Y, et al. Tau activation of microglial cGAS-IFN reduces MEF2C-mediated cognitive resilience. Nat Neurosci. 2023;26:737-50.
258. Xie X, Ma G, Li X, Zhao J, Zhao Z, Zeng J. Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5×FAD mice. Nat Aging. 2023;3:202-12.
259. Chung S, Jeong JH, Park JC, et al. Blockade of STING activation alleviates microglial dysfunction and a broad spectrum of Alzheimer’s disease pathologies. Exp Mol Med. 2024;56:1936-51.
260. Zhou W, Mohr L, Maciejowski J, Kranzusch PJ. cGAS phase separation inhibits TREX1-mediated DNA degradation and enhances cytosolic DNA sensing. Mol Cell. 2021;81:739-755.e7.
261. Lv G, Wang Q, Lin L, et al. mTORC2-driven chromatin cGAS mediates chemoresistance through epigenetic reprogramming in colorectal cancer. Nat Cell Biol. 2024;26:1585-96.
262. Chen X, Holtzman DM. Emerging roles of innate and adaptive immunity in Alzheimer’s disease. Immunity. 2022;55:2236-54.
264. Vandenberghe R, Riviere ME, Caputo A, et al. Active Aβ immunotherapy CAD106 in Alzheimer’s disease: a phase 2b study. Alzheimers Dement. 2017;3:10-22.
265. Riviere ME, Langbaum JB, Turner RS, et al. Effects of the active amyloid beta immunotherapy CAD106 on PET measurements of amyloid plaque deposition in cognitively unimpaired APOE ε4 homozygotes. Alzheimers Dement. 2024;20:1839-50.
266. Novak P, Kovacech B, Katina S, et al. ADAMANT: a placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer’s disease. Nat Aging. 2021;1:521-34.
267. Novak P, Schmidt R, Kontsekova E, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16:123-34.
268. Panza F, Lozupone M, Logroscino G, Imbimbo BP. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15:73-88.
269. Terao I, Kodama W. Comparative efficacy, tolerability and acceptability of donanemab, lecanemab, aducanumab and lithium on cognitive function in mild cognitive impairment and Alzheimer’s disease: a systematic review and network meta-analysis. Ageing Res Rev. 2024;94:102203.
270. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis. 2022;9:197-210.
271. Salloway S, Pain A, Lee E, et al. TRAILBLAZER-ALZ 4: A phase 3 trial comparing donanemab with aducanumab on amyloid plaque clearance in early, symptomatic Alzheimer’s disease. Alzheimers Dement. 2025;21:e70293.
272. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388:9-21.
273. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. 2021;13:80.
274. Honig LS, Barakos J, Dhadda S, et al. ARIA in patients treated with lecanemab (BAN2401) in a phase 2 study in early Alzheimer’s disease. Alzheimers Dement. 2023;9:e12377.
275. Sperling RA, Jack CR Jr, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367-85.
276. Honig LS, Sabbagh MN, van Dyck CH, et al. Updated safety results from phase 3 lecanemab study in early Alzheimer’s disease. Alzheimers Res Ther. 2024;16:105.
277. Wang S, Mustafa M, Yuede CM, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med. 2020:217.
278. Jain N, Lewis CA, Ulrich JD, Holtzman DM. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. J Exp Med. 2023:220.
279. Griciuc A, Patel S, Federico AN, et al. TREM2 acts downstream of CD33 in modulating microglial pathology in Alzheimer’s disease. Neuron. 2019;103:820-835.e7.
280. Arranz AM, De Strooper B. The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol. 2019;18:406-14.
281. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367-402.
282. Venegas C, Kumar S, Franklin BS, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature. 2017;552:355-61.
283. Roy ER, Wang B, Wan YW, et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020;130:1912-30.
284. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50-6.
285. Pannell M, Economopoulos V, Wilson TC, et al. Imaging of translocator protein upregulation is selective for pro-inflammatory polarized astrocytes and microglia. Glia. 2020;68:280-97.
286. Lomoio S, Pandey RS, Rouleau N, et al. 3D bioengineered neural tissue generated from patient-derived iPSCs mimics time-dependent phenotypes and transcriptional features of Alzheimer’s disease. Mol Psychiatry. 2023;28:5390-401.
287. Fattorelli N, Martinez-Muriana A, Wolfs L, Geric I, De Strooper B, Mancuso R. Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat Protoc. 2021;16:1013-33.
288. Mancuso R, Fattorelli N, Martinez-Muriana A, et al. Xenografted human microglia display diverse transcriptomic states in response to Alzheimer’s disease-related amyloid-β pathology. Nat Neurosci. 2024;27:886-900.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].