
A new route for mitigating age-related cognitive decline
 0
0 Abstract
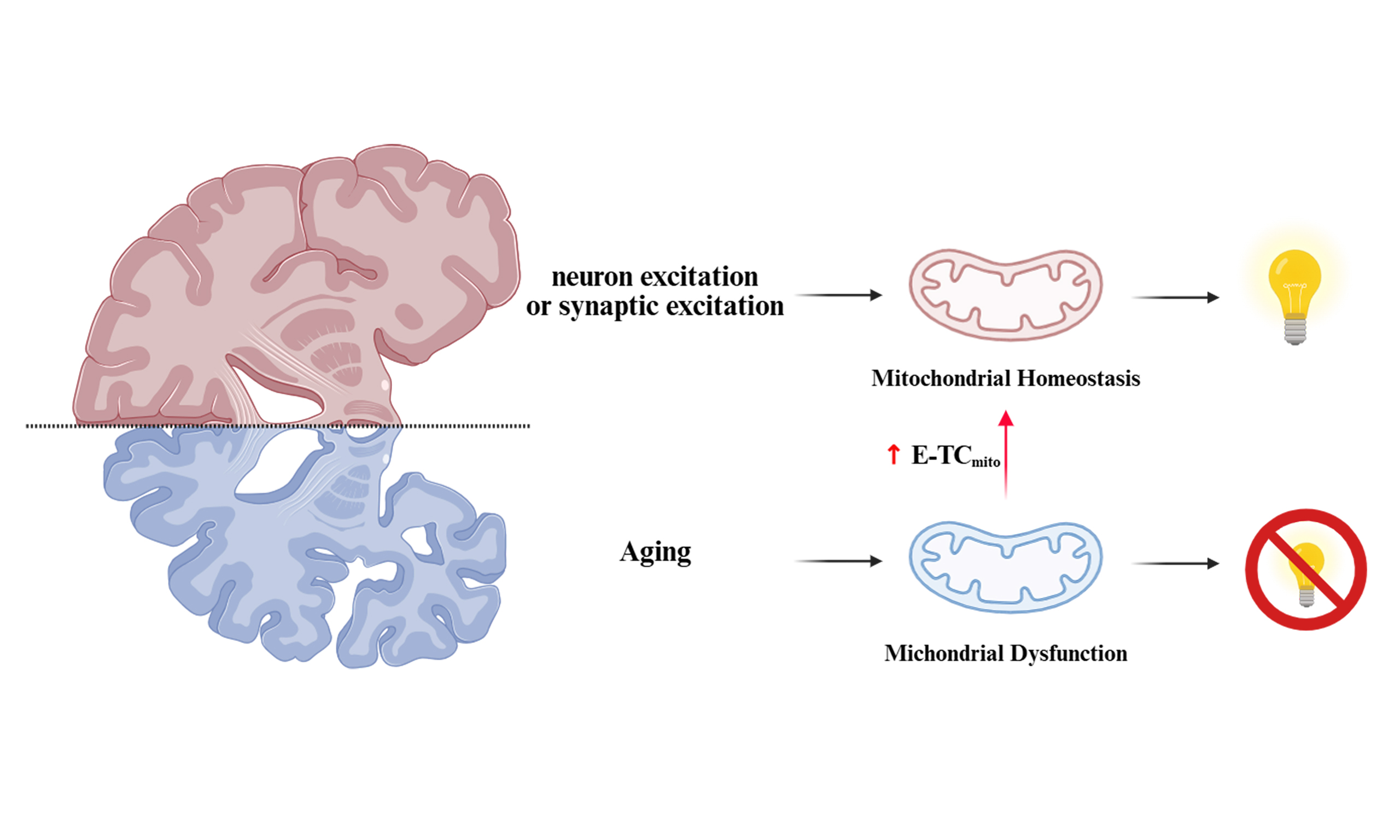
The dynamic relationship between neuronal activity and mitochondrial function plays a crucial role in cognitive health, with disruptions linked to age-related cognitive decline. In this mini review, we highlight the significant findings of Li et al., who discovered an age-dependent coupling mechanism between neuronal/synaptic excitation and excitation-dependent mitochondrial gene transcription coupling (E-TCmito). Their research revealed that enhancing brain E-TCmito in aged animals effectively mitigated age-related cognitive decline.
Keywords
INTRODUCTION
Aging is a primary risk factor for cognitive decline, yet the underlying cellular mechanisms are not yet fully understood. Mitochondrial dysfunction has been implicated, but further research is required to elucidate the mechanisms by which neuronal activity dynamically regulates mitochondrial gene expression to meet energy demands, and how this process changes with age. The prevailing concept of excitation-transcription coupling (E-TC), which establishes a link between neuronal activity and nuclear gene expression, had not been examined in the context of the mitochondrial genome. The seminal work by Li et al. addresses this gap by proposing and validating the existence of mitochondrial E-TC (E-TCmito)[1]. Their research reveals that an age-related failure in activity-dependent calcium signaling within mitochondria impairs this process, leading to synaptic and memory deficits. This introduction provides the necessary context for the discovery of a fundamental regulatory mechanism for neuronal energy homeostasis, and its profound implications for understanding cognitive aging.
MAIN TEXT
Aging inevitably contributes to cognitive decline, though the underlying mechanisms remain under extensive investigation. At the biological level, the aging brain is subject to progressive oxidative stress and neuroinflammation. These age-related pathological processes may interfere with calcium homeostasis, thereby damaging the efficiency of mitochondrial gene expression, indicating a close relationship with mitochondrial dysfunction[1]. While mitochondrial dysfunction is linked to cognitive decline, the age-dependent mechanisms of mitochondrial gene expression remain underexplored. How mitochondria adapt their gene expression to meet neurons’ dynamic energy demands remains not fully understood. A sharper focus on this gap would contextualize the study’s novelty. E-TC describes how neurons modify their transcriptional programs in response to external stimuli[2-4]. In consideration of the distinctive attributes of the mitochondrion - a specialized extranuclear organelle that is endowed with its own genetic system - Li
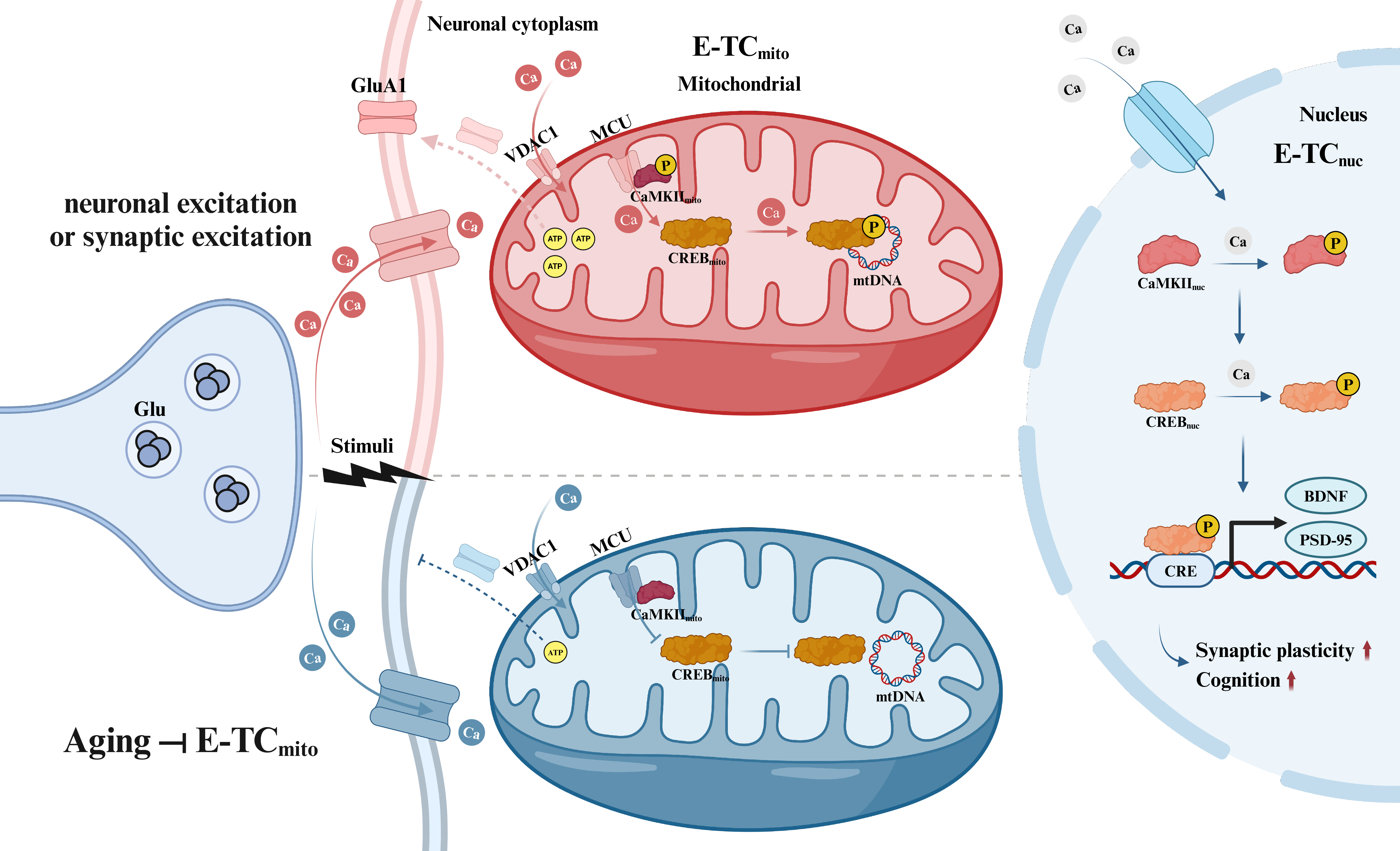
Figure 1. Model of excitation-dependent mitochondrial gene expression (E-TCmito) in aging and young neurons. During periods of neuronal or synaptic excitation, calcium is taken up by mitochondria in neurons. This process activates CaMKII in the mitochondrial membrane and CREB in the mitochondrial matrix (E-TCmito), triggering transcription of mitochondrial DNA for ATP production. The resulting ATP supports surface expression of GluA1 at the synapse. This mechanism is distinct from conventional excitation-dependent nuclear gene transcription (E-TCnuc). Research has demonstrated that aging significantly reduces the efficiency of E-TCmito engagement during mitochondrial calcium uptake, consequently impairing mtDNA transcription, diminishing ATP production, and reducing receptor surface expression [Created in BioRender. Xiaoxing, L. (2025) https://BioRender.com/yz6uzzm]. E-TCmito: Excitation and excitation-dependent mitochondrial gene transcription coupling; CaMKII: calcium–CaM-dependent protein kinase II; CaM: calmodulin; CREB: Ca2+/cAMP response element-binding protein; ATP: adenosine triphosphate; GluA1: glutamate receptor 1; E-TCnuc: excitation-dependent nuclear gene transcription coupling; mtDNA: mitochondrial DNA; VDAC1: voltage-dependent anion channel 1; MCU: mitochondrial calcium uniporter; BDNF: brain-derived neurotrophic factor; PSD-95: postsynaptic density protein 95; CRE: cAMP response element.
Strikingly, Li et al. discovered that mitochondrial calcium influx becomes less efficient at encoding neuronal activity changes with age. Recognizing calcium-dependent signaling’s pivotal role in regulating DNA transcription and the connection between the excitation state of neurons and fluctuations in mitochondrial calcium ([Ca2+]mito) that occur in response to activity, they investigated whether the diminished increase in mtDNA transcription following stimulation in aged mice might relate to alterations in activity-dependent [Ca2+]mito. To investigate this link directly, the researchers created a novel sensor by fusing the green calcium indicator GCaMP6s to a mitochondrial targeting sequence (MTS). This design specifically localizes the sensor, termed Mt-GCaMP6s, to the mitochondrial matrix. Using the mitochondrial-targeted calcium sensor, the researchers measured calcium influx into mitochondria within the primary somatosensory cortex while administering whisker stimulation to both young and aged mice. The researchers observed that this influx was significantly attenuated in older mice, suggesting a mismatch between mitochondrial energetics and neuronal activity during the aging process.
Subsequently, the researchers sought to determine whether calcium signaling is essential for E-TCmito and if impaired calcium signaling affects synaptic plasticity. Their investigation centered on [Ca2+]mito’s role in E-TCmito, using primary neocortical neurons as a model system. Treatment of these neurons with bicuculline, a γ-aminobutyric acid type A (GABAA) receptor antagonist, resulted in an enhancement in both synaptic excitatory transmission and the firing activity of neurons. This treatment increased [Ca2+]mito and elevated mtDNA transcription. Importantly, blocking calcium influx using various reagents inhibited E-TCmito, confirming its dependence on activity-related [Ca2+]mito. In conclusion, Li et al. demonstrated the crucial role of calcium-triggered E-TCmito in regulating mitochondrial gene expression in response to neuronal, synaptic, and cognitive activity. According to their findings, the age-dependent E-TCmito process, which involves the activity-driven regulation of mitochondria, is essential for the maintenance of neuronal energy reserves, the preservation of synaptic resilience, and the support of memory function.
In addition to demonstrating that mitochondria possess activity-dependent calcium signaling mechanisms for neuronal and synaptic activity, Li et al. also elucidated the primary signaling cascades that orchestrate the transcription of mtDNA in an activity-dependent manner, providing a possible pathway for the decoding of calcium signals within mitochondria.
In a crucial series of experiments, Li et al. demonstrated that neuronal and synaptic excitation initiated calcium signaling, activating calcium–calmodulin (CaM)-dependent protein kinase II (CaMKII) in the mitochondrial membrane and Ca2+/cAMP response element-binding protein (CREB) in the mitochondrial matrix. It was discovered that CREB, a mitochondrial transcription factor recruited by neuronal activity, plays a critical role in enabling neurons to adapt during the processes of upscaling and downscaling. When mitochondrial CREB (CREBmito) was inhibited in preparations of excitatory neuron cultures, it impaired mitochondrial gene expression and reduced excitatory synaptic current strength, along with a reduction in the surface expression of the glutamate receptor glutamate receptor 1 (GluA1) subunit, which was observed after drug-induced increases in network activity. Consequently, even though neurons exhibited a more rapid response to sustained alterations in synaptic transmission, they took a longer time to revert to the baseline state following the withdrawal of the stimulus. To further explore the importance of CREB, researchers employed a genetic modification technique to inhibit CREB expression in excitatory neurons in old mice. They revealed that the mice exhibited notable impairments in behavioral experiments, including the contextual fear conditioning (CFC) task and the novel location recognition task. Additionally, researchers observed reduced expression levels of mitochondria-related genes in these subjects. Importantly, both the behavioral performance and gene expression levels were restored following CREB re-expression, suggesting potential therapeutic interventions targeting CREB for related neurological conditions.
It is noteworthy that CaMKII, the most abundant protein in excitatory synapses and a crucial regulator of synaptic plasticity, learning, and memory[6], has received limited attention in mitochondrial research. This kinase’s activation begins with increased intracellular calcium levels, triggering molecular cascades essential for synaptic plasticity[7]. CaMKII phosphorylates numerous synaptic proteins, modulating their structure and function. These processes connect intimately with molecular events critical for synaptic plasticity, including receptor trafficking, localization, and activity; actin cytoskeletal dynamics; translation; and even transcription through synapse-nucleus shuttling[6]. Li et al. have uncovered a significant mitochondrial role for this element, providing novel insights into CaMKII’s function in E-TCmito.
Furthermore, CREB, a calcium-activated protein in decoding calcium-induced changes in CaMKII to influence synaptic plasticity, has been primarily studied in the nucleus[8]. Mechanistically, CREB’s role in memory is believed to stem from its involvement in long-term synaptic plasticity[9]. This process involves transmitting events from cell membranes into gene expression alterations, with stronger validation in the nucleus[10]. Li et al. have demonstrated that CREB, beyond its nuclear role, also modulates the intrinsic excitability of the mitochondrial apparatus. This establishes novel and compelling connections between neuronal and synaptic excitation and E-TCmito, significantly advancing our understanding of CREB’s role in E-TCmito. Undoubtedly, some issues persist that necessitate further in-depth investigation. Specifically, how can the regulators of CREB/CaMKII be precisely targeted and effectively delivered to the mitochondria of specific neurons? Additionally, given that E-TCmito and certain cellular processes share some common signaling pathways, could its long-term activation potentially result in elevated oxidative stress? The opportunity to explore these questions, resting on the fundamental mechanisms elucidated by Li et al., suggests that a paradigm shift is imminent in the study of mitochondrial aging, opening up new avenues for exploration.
In sum, Li et al. present a comprehensive analysis of mitochondrial biology in the aging mammalian brain. Through innovative tools, sophisticated physiological mechanisms, and compelling behavioral experiments, they provide fundamental insights into the complex processes underlying age-related cognitive changes. Their findings suggest that the identified factors may serve as viable therapeutic targets for age-related cognitive decline, including Alzheimer’s and Parkinson’s diseases[11]. The researchers propose a hypothesis illuminating potential mechanisms through which aging processes might be arrested, potentially preventing age-related disease development.
The study’s discoveries regarding mtDNA transcription in neurons carry profound implications for future research in mitochondrial metabolic disease[12,13]. The identification of CaMKII and CREB as responders to calcium signaling changes in mitochondria represents a novel finding that opens new research avenues. Furthermore, this work provides a foundation for investigating potential analogous mechanisms of mitochondrial excitatory transcription coupling in non-neuronal cells, which could influence metabolic diseases in other systems. Overall, these novel findings and future directions establish a basis for advancing our understanding of age-related cognitive decline and developing innovative therapeutic strategies to enhance elderly individuals’ quality of life.
CONCLUSION
In conclusion, the work by Li et al. fundamentally advances our understanding of brain aging by elucidating a novel mechanism of E-TCmito. The research demonstrates that the age-related breakdown of activity-dependent calcium signaling to the mitochondrion, and the consequent failure to activate the intra-mitochondrial CaMKII-CREB axis, is a critical factor in synaptic dysfunction and memory decline[5]. The present study proposes a novel framework for understanding age-related cognitive deficits by bridging the gap between neuronal activity, mitochondrial gene expression, and cognitive output. In addition, it firmly establishes mitochondrial E-TCmito and its key regulators as promising therapeutic targets for combating neurodegenerative diseases.
DECLARATIONS
Acknowledgments
We thank Xiaoxing Liu for assistance with Figure 1 and Graphic Abstract creation using BioRender.com [Graphic Abstract was created in BioRender. Xiaoxing, L. (2025) https://BioRender.com/fm69lbe].
Authors’ contributions
Wrote the first draft of the mini review: Chen H
Edited and contributed to the final draft: Cao L, Liu S, Yuan K
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Key Research and Development Program of China (2023YFC2506800), the National Natural Science Foundation of China (No. 82371499), and the Young Elite Scientists Sponsorship Program by CAST (2023QNRC001).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Kaliszewska A, Allison J, Martini M, Arias N. Improving age-related cognitive decline through dietary interventions targeting mitochondrial dysfunction. Int J Mol Sci. 2021;22:3574.
2. Morgan JI, Curran T. Role of ion flux in the control of c-fos expression. Nature. 1986;322:552-5.
3. Greenberg ME, Ziff EB, Greene LA. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science. 1986;234:80-3.
4. West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat Rev Neurosci. 2002;3:921-31.
5. Li W, Li J, Li J, et al. Boosting neuronal activity-driven mitochondrial DNA transcription improves cognition in aged mice. Science. 2024;386:eadp6547.
6. Yasuda R, Hayashi Y, Hell JW. CaMKII: a central molecular organizer of synaptic plasticity, learning and memory. Nat Rev Neurosci. 2022;23:666-82.
8. Ma H, Groth RD, Cohen SM, et al. γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell. 2014;159:281-94.
9. Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230-40.
10. Parra-Damas A, Saura CA. Synapse-to-nucleus signaling in neurodegenerative and neuropsychiatric disorders. Biol Psychiatry. 2019;86:87-96.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].