


Non-coding RNA: emerging players and therapeutic targets in heart failure
 0
0 Abstract
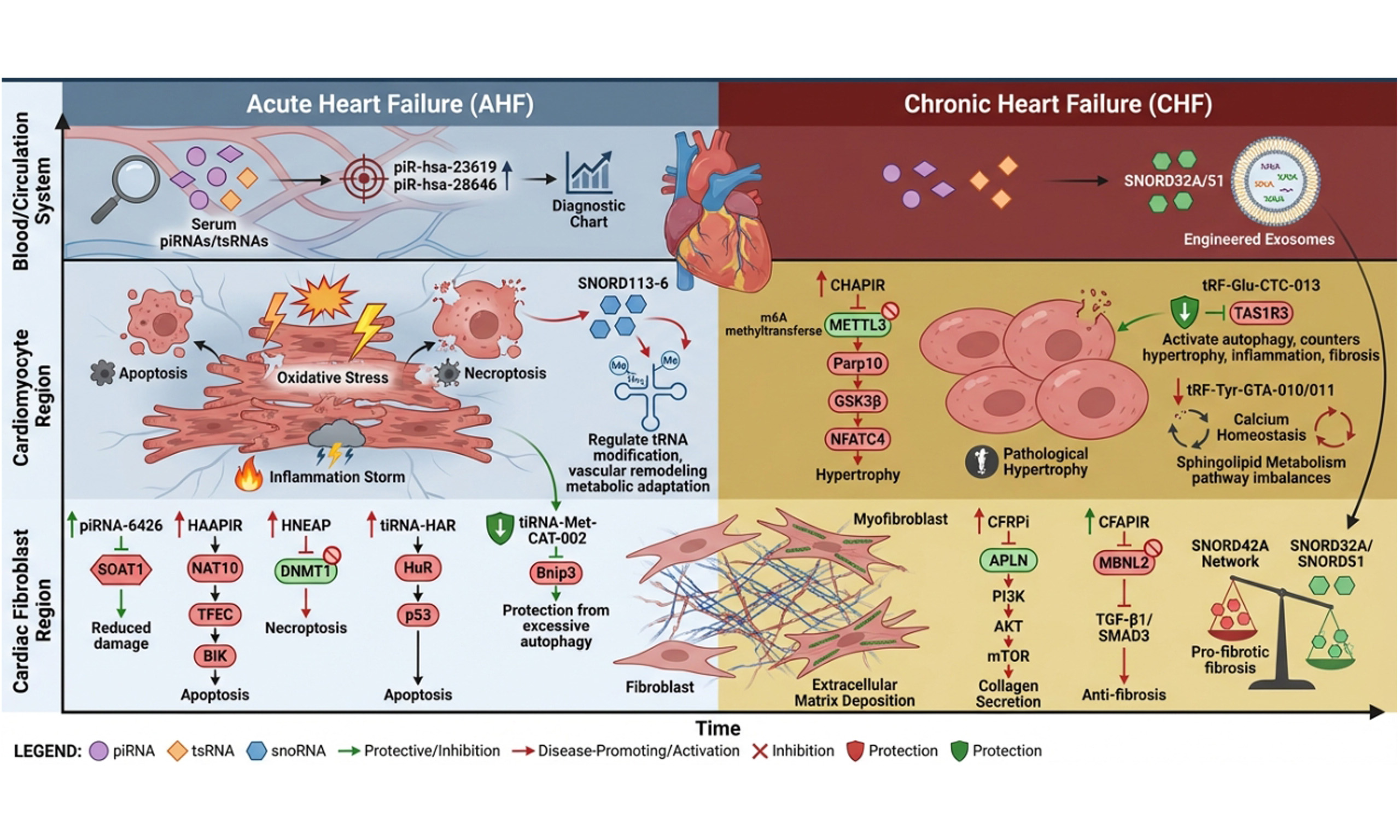
Heart failure (HF) is a syndrome of global concern with high morbidity and mortality, whose complex molecular regulatory mechanisms are not yet fully understood. Moving beyond the traditional research framework focused on microRNAs, long non-coding RNAs (ncRNAs), and circular RNAs, this review concentrates on the pivotal roles of emerging ncRNAs - specifically Piwi-interacting RNAs (piRNAs), transfer RNA-derived small RNAs (tsRNAs), and small nucleolar RNAs (snoRNAs) - in the pathological progression of HF. In the acute phase of HF, these molecules rapidly respond to stressors such as ischemia and hypoxia. They directly influence cardiomyocyte fate and acute injury outcomes by regulating processes including apoptosis, necroptosis, autophagy, and inflammatory responses. During the chronic phase, they are deeply involved in pathological myocardial remodeling. They precisely regulate cardiomyocyte hypertrophy, cardiac fibroblast activation, and interstitial fibrosis in a cell-specific manner, maintaining a fine-tuned balance between pro-pathological and protective functions. These discoveries significantly enrich the molecular regulatory map of HF and reveal the considerable potential of these ncRNAs as novel non-invasive biomarkers and promising therapeutic targets. Of particular note, strategies employing engineered exosomes to deliver specific snoRNAs have demonstrated therapeutic effects in preclinical models, such as reversing fibrosis and improving cardiac function. This marks a shift in the treatment paradigm for HF toward precise RNA-level regulation.
Keywords
INTRODUCTION
Heart failure (HF) is a heterogeneous syndrome characterized by impaired cardiac pumping or congestion capacity. It is a leading cause of hospitalization and mortality worldwide, affecting over 64 million people globally[1]. Physiologically, HF can be defined as either inadequate cardiac output to meet metabolic demands secondary to compensatory neurohormonal activation, or as adequate cardiac output (often manifested by elevated left ventricular filling pressure) secondary to compensatory neurohormonal activation[2]. Furthermore, HF can develop from multiple pathological conditions, including myocardial infarction (MI), pressure overload (aortic stenosis and hypertension), inflammatory cardiomyopathies (myocarditis), and volume overload (valvular regurgitation)[3].
Based on the acute or chronic onset of HF, it can be classified as chronic (CHF) or acute heart failure (AHF)[4]. CHF is rare in young adults but common in the elderly, who account for 80% of patients with this syndrome. Its incidence and prevalence increase with age[5]. CHF has several underlying causes (ischemic disease, dilated cardiomyopathy, hypertrophic cardiomyopathy, hypertension, valvular heart disease, etc.), leading to alterations in cardiac structure (left ventricular hypertrophy or dilation, cardiac fibrosis, cardiac remodeling) and function (abnormal cardiac output, diastolic dysfunction), resulting in common symptoms[6]. AHF is broadly defined as the rapid onset or worsening of physical signs and symptoms of HF[7]. This umbrella term encompasses patients experiencing the first occurrence of classic HF symptoms and signs (new-onset AHF) as well as those with worsening pre-existing cardiomyopathy (acute decompensated HF). Symptoms include dyspnea, fatigue, pulmonary crackles, peripheral edema, and jugular venous distension on physical examination[8].
At the molecular level, HF is accompanied by an imbalance in cardiac homeostasis, a process precisely regulated by multiple types of RNA molecules. In the cardiovascular field, non-coding RNAs (ncRNAs) have emerged as crucial regulatory molecules, participating in cardiac physiological functions and pathological processes through complex mechanisms[5]. ncRNAs are a class of RNAs that do not serve as templates for translating proteins. They participate in regulating messenger RNA (mRNA) translation, RNA splicing, DNA replication and repair, gene transcription, development, and cell differentiation[5]. Based on their biological functions, ncRNAs can be broadly categorized into two classes: housekeeping ncRNAs and regulatory ncRNAs. Among these, housekeeping ncRNAs include ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNAs (snRNA), and small nucleolar RNAs (snoRNA)[5]. Based on the length of the transcribed product, ncRNAs with regulatory functions can be subdivided into short ncRNAs and long ncRNAs (lncRNAs). Short ncRNAs include Piwi-interacting RNAs (piRNAs), microRNAs (miRNAs), and small-interfering RNAs (siRNAs). lncRNAs are a class of ncRNAs lacking 100 amino acids and exceeding 200 nucleotides in length, exhibiting complex and diverse structures and functions[9]. Additionally, linear products (miRNAs, lncRNAs) and circular RNAs (circRNAs) can be classified based on the linearity or circularity of the transcribed products[10]. Currently, miRNAs, lncRNAs, and circRNAs have been demonstrated to play roles in HF. Beyond these, piRNAs, comprising 26-31 nucleotides, interact with the Piwi subfamily of Argonaute proteins to form RNA-induced silencing complex and function in transposon silencing[11,12].
Studies indicate that piRNAs exert cell-specific regulatory effects in myocardial hypertrophy and cardiac interstitial fibrosis, and serve as potential targets for myocardial injury in AHF. tRNA-derived small RNAs (tsRNAs) can be categorized into two classes[13]: tRNA-derived fragments (tRFs) with a length of approximately 14-30 nt, which are further divided into four subclasses - tRF-1, tRF-3, tRF-5, and i-tRF; and tRNA-derived stress-induced RNAs (tiRNAs) with a length of 28-36 nt[14]. Among these, tRFs regulate calcium ion transport to maintain myocardial electrophysiological homeostasis and suppress arrhythmias[15]. tRFs possess multiple functions, including regulating protein translation, silencing mRNA, and modulating transposon activity, and may play a role in cardiac hypertrophy[16,17]. tiRNAs are slightly longer than tRFs, typically ranging from 31 to 41 nucleotides in length; they are also referred to as “tRNA halves” which promote cardiac activities such as cardiomyocyte apoptosis (CA) by transmitting signal[18]. snoRNAs are slightly longer transcripts, ranging from 60 to 300 nucleotides in length, believed to mediate chemical modifications of other RNA transcripts[19]. This facilitates rRNA processing but may also influence mRNA splicing[20].
Although current research has made some progress in treating HF, mortality remains exceptionally high due to insufficient understanding of its cellular and molecular causes, representing an unmet medical need worldwide[21,22]. This review aims to transcend the traditional miRNA/lncRNA/circRNA research framework, focusing instead on these “non-mainstream” ncRNAs. This article systematically reviews the latest research evidence on molecules such as piRNAs and snoRNAs in HF, explores their potential as novel biomarkers, and discusses their prospects for application in HF gene therapy. By summarizing current research gaps and challenges, this review aims to provide new perspectives and directions for a comprehensive understanding of the molecular regulatory network in HF and the development of innovative diagnostic and therapeutic strategies.
AHF AND NCRNAS
piRNA
AHF, often triggered by acute myocardial infarction (AMI) and ischemia/reperfusion (I/R) injury, is characterized by core pathological processes including rapid myocardial cell death (apoptosis and necroptosis), intense oxidative stress, and inflammatory responses[23]. Recent studies indicate that piRNAs act as critical regulators in this process, and their dysregulated expression directly contributes to the pathogenesis of AHF[24].
piRNA-6426
Sequencing data revealed that piRNA-6426 expression was downregulated in the blood of HF patients compared to healthy individual[25]. Overexpression of piRNA-6426 significantly enhances cell viability and glucose uptake in hypoxia-induced cardiomyocytes, while effectively reducing apoptosis rates, reactive oxygen species (ROS) production, lactate dehydrogenase (LDH) activity, and levels of inflammatory cytokines Interleukin-1 beta (IL-1β) and Tumor Necrosis Factor-alpha (TNF-α). Further mechanistic studies revealed that piRNA-6426 increases the methylation level of the promoter region of the cholesterol acyltransferase 1 (SOAT1) gene by promoting the enrichment of DNA methyltransferase 3B (DNMT3B), thereby suppressing SOAT1 expression. This epigenetic regulatory mechanism ultimately reduced hypoxia-induced oxidative stress and inflammatory responses, alleviating cellular dysfunction. The in vivo efficacy of this function was validated in a rat HF model: following injection of a piRNA-6426-overexpressing lentiviral vector into HF rats, both cardiac inflammation and health indices showed significant improvement[26]. Thus, downregulation of piRNA-6426 may directly contribute to the pathogenesis of HF, and its overexpression holds promise as a therapeutic strategy to alleviate HF-associated myocardial injury and dysfunction.
Heart apoptosis-associated piRNA and heart necroptosis-associated piRNA
Beyond the protective role of piRNA-6426, the study further identified two pathogenic piRNAs, each predominantly responsible for a distinct mode of cell death. Among these, the heart apoptosis-associated piRNA (HAAPIR) was confirmed as a key regulator of CA in response to I/R injury. HAAPIR deficiency mitigates I/R-induced MI and improves cardiac function. Mechanistically, HAAPIR directly interacts with N4-acetylcytidylyltransferase NAT10, enhancing its activity to promote ac4C acetylation modification of transcription factor EC (TFEC) mRNA, thereby upregulating TFEC protein expression. As a transcription factor, TFEC further activates the transcription of the pro-apoptotic gene BCL2 (B cell lymphoma 2)-interacting killer factor (BIK), ultimately driving the CA process. Thus, the HAAPIR-NAT10-TFEC-BIK signaling axis constitutes a distinct pro-apoptotic pathway that may serve as a potential target for reducing myocardial injury caused by CA in ischemic heart disease[27] [Figure 1A].
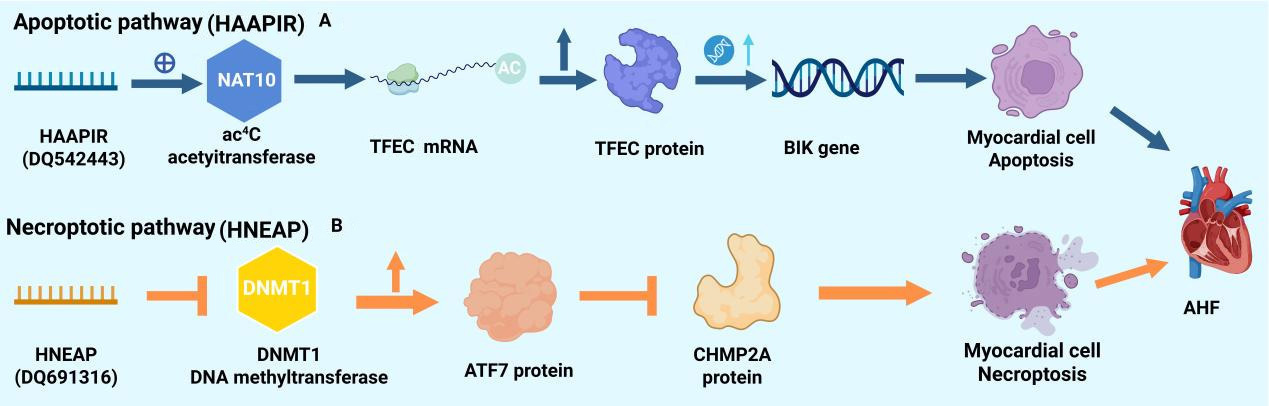
Figure 1. Dual regulatory pathways of programmed cell death in cardiomyocytes mediated by piRNAs. (A) HAAPIR-mediated apoptotic pathway. In response to cellular stress, the piRNA HAAPIR upregulates the expression of the acetyltransferase NAT10. NAT10 catalyzes ac4C acetylation on TFEC mRNA, enhancing its stability and translation, leading to increased TFEC protein levels. TFEC, as a transcription factor, subsequently activates the expression of the pro-apoptotic gene BIK, ultimately inducing CA; (B) HNEAP-mediated necroptotic pathway. The piRNA HNEAP promotes the expression of the DNA methyltransferase DNMT1. Elevated DNMT1 levels lead to increased production of the transcription factor ATF7. ATF7 upregulates target genes that suppress the expression of CHMP2A, a key inhibitor of the necroptotic pathway. The loss of CHMP2A inhibition thereby predisposes cardiomyocytes to necroptosis. Created with BioRender.com. (2026) https://BioRender.com/0583atk. piRNAs: Piwi-interacting RNAs; CA: cardiomyocyte apoptosis; AHF: acute heart failure; BIK: BCL2 (B cell lymphoma 2)-interacting killer factor; mRNA: messenger RNA; TFEC: transcription factor EC.
In parallel with apoptosis, necroptosis represents another critical pathway in AMI. A study identified a significantly upregulated piRNA - DQ691316 - in hypoxia/reoxygenation (H/R)-injured cardiomyocytes, termed the heart necroptosis-associated piRNA (HNEAP). Primarily expressed in cardiomyocytes compared to cardiac fibroblasts, HNEAP suppresses the RNA 5-methylcytosine (m5C) methylation activity mediated by DNA methyltransferase 1 (DNMT1) through direct interaction with this enzyme. This action upregulates transcription factor ATF7, which antagonizes the necroptosis-inhibiting factor Charged multivesicular body protein 2A (CHMP2A), ultimately promoting cardiomyocyte necroptosis. Targeting HNEAP or ATF7 knockdown effectively mitigated pathological stimulus-induced myocardial injury, highlighting the pathway’s potential as a therapeutic target. Thus, HNEAP may represent a promising target for reducing infarct size and improving cardiac function following I/R injury. The discovery of these pro-apoptotic and pro-necroptotic piRNAs suggests that targeting HAAPIR or HNEAP may help reduce myocardial remodeling and HF progression caused by cardiomyocyte death, particularly in the context of ischemic heart disease[28] [Figure 1B].
Serum piRNAs
From a clinical translational perspective, high-throughput sequencing analysis revealed extensive piRNA expression dysregulation in the serum of AMI patients: 195 piRNAs were upregulated and 13 piRNAs were downregulated. Validation via quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) identified piR-hsa-9010, piR-hsa-28646, and piR-hsa-23619 as the most significantly upregulated piRNAs[29]. Bioinformatics analysis suggested that piR-hsa-23619 may exacerbate post-MI inflammatory responses by participating in the tumor necrosis factor (TNF) signaling pathway, thereby affecting vascular function after MI. Meanwhile, piR-hsa-28646 may influence angiogenesis and repair processes by regulating the Wnt signaling pathway(also known as the Wnt/β-catenin signaling pathway). More importantly, compared to mRNA, piRNAs exhibit higher stability in blood, making them highly attractive novel non-invasive biomarkers for early diagnosis and risk assessment of AMI. Therefore, the altered expression of these serum piRNAs holds diagnostic potential for AMI, offering new avenues for early prevention and management of HF. However, large-scale validation is still required before they can serve as noninvasive markers for predicting the risk of HF following AMI[30].
tsRNA
Unlike piRNA, tsRNA has been demonstrated to play a pivotal regulatory role in CA, necroptosis, autophagy imbalance, and acute inflammatory responses[31].
Hypoxia responsive-tiRNA
Wang et al. discovered that in MI mouse models and hypoxic cardiomyocytes, expression of hypoxia responsive-tiRNA (tiRNA-HAR) (AS-tDR-001449) - a 5’-end fragment derived from tRNA-Val-CAC [stands for transfer RNA that carries valine (Val) and possesses the anticodon sequence CAC] - was significantly upregulated[32]. Further mechanistic investigation revealed that hypoxia induced Hypoxia Inducible Factor 1α (HIF1α) protein expression, which in turn elevated Angiogenin (ANG) mRNA and protein levels. Inhibiting HIF1α or silencing ANG reversed hypoxia-induced tiRNA-HAR upregulation, confirming its expression regulation by the HIF1α/ANG axis[33]. Functionally, tiRNA-HAR overexpression significantly enhanced Cleaved-caspase3 expression and increased TdT-mediated dUTP Nick-End Labeling (TUNEL)-positive cell counts under both normoxic and hypoxic conditions, promoting CA. Conversely, Antisense Oligonucleotide (ASO)-mediated silencing of tiRNA-HAR markedly suppressed hypoxia-induced apoptosis and Cleaved-caspase3 expression. At the animal level, tail vein injection of Adeno-Associated Virus 9 (AAV9)-ASO significantly reduced tiRNA-HAR levels in the myocardium of MI mice and improved cardiac function: At 3 days and 4 weeks post-MI, the silenced group exhibited significantly higher left ventricular ejection fraction (EF%) and short-axis shortening fraction (FS%) compared to the control group. Concurrently, myocardial apoptosis (CA), fibrotic area, and expression of remodeling-related genes [ANP, Collagen Type I Alpha 1 Chain (COL1A1), etc.] were markedly reduced. Regarding molecular mechanisms, tiRNA-HAR significantly elevated p53 mRNA and protein levels. It enhanced p53 mRNA stability by binding Human Antigen R (HuR) and strengthening its interaction with p53 mRNA, as confirmed by actinomycin D experiments. In summary, this study reveals that myocardial ischemia/ hypoxia induces tiRNA-HAR expression upregulation via the HIF1α/ANG axis[32]. tiRNA-HAR directly binds HuR and enhances its binding capacity with p53 mRNA, thereby increasing p53 mRNA stability and ultimately promoting CA. Intervening tiRNA-HAR effectively alleviates myocardial injury and improves cardiac function, providing new theoretical basis for mechanism research and therapeutic strategy development in ischemic heart disease[32].
tiRNA-met-CAT-002
Contrary to the pro-injury effects of tiRNA-HAR, Deng et al. found that tiRNA-Met-CAT-002 expression was induced and upregulated via the HIF1α/ANG axis in a myocardial I/R injury model[34]. Sequencing identified 323 differentially expressed tsRNAs, with 115 showing significant differences (53 upregulated, 62 downregulated). qRT-PCR validated 6 core tsRNAs, among which tiRNA-Met-CAT-002 exhibited the greatest upregulation. Both I/R and H/R induced elevated HIF1α and ANG protein expression. Silencing ANG via siRNA or treating with the HIF1α inhibitor LW6 suppressed tiRNA-Met-CAT-002 upregulation, indicating its expression is regulated by the “hypoxia - HIF1α activation - ANG expression - tiRNA generation” signaling axis. Further TargetScan prediction and experimental validation revealed that the seed sequence of tiRNA-Met-CAT-002 binds to the 3’untranslated region (3’UTR) region of Bnip3 mRNA. A luciferase reporter assay confirmed that this tiRNA significantly suppressed luciferase activity in vectors containing the wild-type Bnip3 3’UTR, while having no effect on mutant vectors. Functionally, tiRNA-Met-CAT-002 suppresses excessive autophagy by targeting Bnip3: under H/R stress, its analog reduced the Microtubule-associated protein 1 light chain 3B isoform II/Microtubule-associated protein 1 light chain 3B isoform I (LC3B-II/LC3B-I) ratio and Beclin-1 expression, increased Sequestosome-1 (p62) accumulation, and diminished autophagosome and autophagy-related spot formation; these effects disappeared upon Bnip3 silencing, indicating that tiRNA-Met-CAT-002 suppresses autophagy flux by negatively regulating Bnip3, thereby enhancing cell viability. In summary, myocardial I/R injury induces tiRNA-Met-CAT-002 upregulation via the HIF1α/ANG axis. This tiRNA directly binds the 3’UTR of Bnip3 and suppresses its expression, thereby mitigating excessive autophagy in cardiomyocytes, enhancing cell viability, and ultimately exerting protective effects against I/R injury. Given that excessive autophagy is a key driver of myocardial remodeling and HF progression, the upregulation of tiRNA-Met-CAT-002 as an endogenous protective mechanism may help limit the transition from acute injury to HF, offering a novel intervention strategy for HF prevention[34,35].
tsRNA as a diagnostic biomarker and treatment heterogeneity indicator for AMI
Su et al. found that many AMI patients with HF experienced no cardiac benefit after sacubitril/valsartan therapy[36]. Through a “sequencing screening - validation diagnosis - Mechanism Exploration” approach, they systematically revealed the pivotal role of tsRNAs. High-throughput sequencing identified 57 significantly differentially expressed tsRNAs between the SVR group (treatment-resistant) and NSVR group (non-treatment-resistant), comprising 36 upregulated and 21 downregulated transcripts. Receiver Operating Characteristic (ROC) curve analysis of three core tsRNAs for diagnosing “sacubitril/valsartan treatment heterogeneity” demonstrated high diagnostic value [Area Under Curve (AUC) > 0.8 for all], positioning them as potential molecular markers for clinically screening SVR patients. Enrichment analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways revealed that tRF-60:76-Val-AAC-1-M5 may target Tumor Necrosis Factor Receptor Superfamily Member 10b (Tnfrsf10b) and BCL2 Like 1 (Bcl2l1), influencing this therapeutic heterogeneity through lipid and atherosclerosis signaling pathways. Furthermore, Gene Ontology (GO) enrichment analysis revealed that target genes of up-regulated tsRNAs primarily participated in “myocardial cell action potential depolarization” and “calcium ion/sodium ion transmembrane transport” (related to myocardial electrophysiological homeostasis). Downregulated tsRNA target genes were primarily involved in “angiogenesis” (related to myocardial perfusion repair after AMI), reflecting the pathological characteristics of “myocardial electrophysiological disorder + insufficient vascular repair” in SVR patients at the functional level[36]. This study directly demonstrates that specific tsRNA profiles not only serve as predictors of differential responses to standard therapies (e.g., sacubitril/valsartan) in HF patients following AMI, but also reveal underlying molecular pathways implicating intrinsic mechanisms driving treatment resistance and refractory HF. This provides critical molecular evidence for achieving precise classification and personalized treatment of HF[37].
snoRNA
Research indicates that snoRNA clusters located at the 14q32 locus play a pivotal role in acute hypoxia stress, oxidative damage, and early tissue remodeling. These clusters not only form the basis of genetic susceptibility but also function as critical stress response molecules involved in the pathological regulation of AHF[38].
Genetic basis and molecular characteristics of the 14q32 snoRNA clusters
Genome-wide association studies reveal a significant association between the 14q32 locus and HF risk (P = 0.018), with this region exhibiting the highest density of snoRNA clusters among single nucleotide polymorphisms. Expression profiling further demonstrates widespread expression of human 14q32 snoRNAs across vascular tissues, exhibiting vascular type specificity. Mechanistically, RNA immunoprecipitation experiments confirmed that these snoRNAs primarily mediate 2’-O-ribosyl methylation by binding to the methyltransferase Fibrillarin. However, their targets may extend to non-canonical mRNA or miRNA precursors, suggesting novel regulatory functions in HF[39]. Future studies may delve into their non-canonical RNA target networks and regulatory mechanisms in HF.
SNORD113-6: coordinating vascular remodeling under acute ischemic stress
As a key member of the 14q32 clusters, Small nucleolar RNA, C/D box 113-6 (SNORD113-6) is significantly upregulated under hypoxia and oxidative stress. By specifically methylating the anticodon region of tRNALeu(TAA) [is transfer RNA that carries leucine (Leu) and possesses the anticodon sequence TAA], SNORD113-6 prevents the cleavage of this tRNA into the tRFLeu47-64 fragment, which promotes vascular remodeling under stress conditions. This theoretically inhibits excessive vascular smooth muscle proliferation, exerting an endogenous protective effect. However, in actual pathological processes, this protective mechanism may manifest as “compensatory failure”. The consequences are twofold: on one hand, unchecked vascular remodeling exacerbates coronary resistance and cardiac afterload; on the other, dysregulated SNORD113-6 may amplify inflammatory cascades, promoting myocardial fibrosis (MF). Together, these factors lead to reduced ventricular compliance and impaired pumping function, creating a vicious cycle[40,41]. Future studies should further evaluate its potential as a therapeutic target in HF and its specific regulatory pathways.
SNORD97/SNORD133: a modifying axis regulating myocardial cell metabolic adaptation
Beyond SNORD113-6, SNORD97 and SNORD133 within the same cluster jointly form a modification network regulating metabolic adaptation in cardiomyocytes. Both molecules synergistically methylate the wobble site (C34) of tRNAMet(CAT) [transfer RNA that carries methionine (Met) and possesses the anticodon sequence CAT], maintaining tRNA stability under stress conditions and thereby inhibiting its cleavage into abnormal tRF fragments. Dysregulation of this network during AHF may trigger tRNA metabolic disorders and protein synthesis impairment, leading to failure of key energy pathways such as fatty acid oxidation in cardiomyocytes. This exacerbates preexisting energy supply crises and ultimately accelerates rapid decompensation of myocardial contractile function[41].
Extracellular vesicle-contained snoRNAs: remote transmission and amplification of injury signals
In the pathological environment of AHF, snoRNAs also serve as crucial intercellular communication messengers. Under ischemic stress, cardiomyocytes and coronary vascular cells can selectively package and secrete specific snoRNAs (such as SNORD96A and SNORD73A) via extracellular vesicles (EVs). Upon being taken up by remote or neighboring coronary endothelial cells and fibroblasts, these snoRNAs enriched in EVs induce an inflammatory and proliferative phenotype in the recipient cells. This process transmits and amplifies local ischemic injury signals, further exacerbating coronary stenosis and MF progression[41].
CHF AND NCRNAS
piRNA
The core pathological basis of CHF is pathological myocardial remodeling, primarily manifested as myocardial cell hypertrophy and cardiac interstitial fibrosis[42]. Recent studies indicate that piRNAs play a crucial cell-specific regulatory role in this chronic progressive process, encompassing both disease-driving “bad molecules” and protective “good molecules”[43].
CHAPIR: key PiRNA driving myocardial hypertrophy
Gao et al. identified CHAPIR (DQ726659) as a key inducer of pathological cardiac hypertrophy using a transcatheter aortic coarctation (TAC) model[44]. It exhibits high tissue-specific expression in the heart, with significantly higher levels in cardiomyocytes than in cardiac fibroblasts. In TAC-induced hypertrophic tissue, CHAPIR expression was markedly elevated, suggesting its involvement in pathological hypertrophy development. Using angiotensin II (Ang II) stimulation of neonatal mouse primary cardiomyocytes to mimic hypertrophic responses, the researchers further validated CHAPIR’s function. At the cellular level, silencing CHAPIR suppressed Ang II-induced increases in cardiomyocyte area and the upregulation of hypertrophy marker genes such as Atrial Natriuretic Peptide (ANP), Brain Natriuretic Peptide (BNP), and Myosin Heavy Chain 7 (Myh7). Functionally, CHAPIR knockout effectively reduced TAC-induced cardiac hypertrophy and fibrosis while improving left ventricular function. At the mechanistic level, CHAPIR forms a complex by binding to the P-element induced wimpy testis-like protein 4 (PIWIL4) protein, thereby interfering with the activity of the N6-Methyladenosine (m6A) Methyltransferase 3, N6-Adenosine-Methyltransferase Complex Catalytic Subunit (METTL3). This interaction does not alter METTL3 expression levels but inhibits its methyltransferase activity, ultimately leading to a reduction in overall m6A levels in cardiomyocytes. Methylated RNA Immunoprecipitation Sequencing (MeRIP-seq) analysis further revealed that poly ADP-ribose polymerase 10 (Parap10) mRNA is a specific target of CHAPIR. Reduced m6A modification enhances mRNA stability, leading to increased Parp10 protein expression. The elevated Parp10 inhibits Glycogen Synthase Kinase-3 (GSK3β) kinase activity through mono-ADP-ribosylation. The decreased GSK3β activity weakens its phosphorylation of the transcription factor Nuclear factor of activated T cells 4 (NFATC4), thereby inhibiting NFATC4 nuclear export. This leads to NFATC4 accumulation within the nucleus, ultimately activating the transcription of hypertrophy-related genes such as Anp and Myh7, driving the process of pathological cardiac hypertrophy[45].
The core pathological basis of CHF is myocardial remodeling, including changes such as hypertrophy and fibrosis. This study identifies CHAPIR as a key molecular driver of this process. Combined with previous reports indicating the diagnostic potential of piRNAs in AMI, these findings suggest that piRNAs may exert stage-specific regulatory roles across different phases of cardiovascular disease - from acute ischemic injury and chronic myocardial remodeling to HF - carrying significant pathological and translational implications[44].
Cardiac fibrosis-associated piRNA and cardiacfibrosis associated piRNA: antagonistic roles in cardiac fibrosis
Unlike CHAPIR, which primarily acts on cardiomyocytes, the core of the fibrosis process lies in the abnormal activation of cardiac fibroblasts[46]. Research reveals that two functionally opposing piRNAs form an intricate regulatory network in this process.
Among various cardiac cell types isolated from normal mice, piRNA-000691 exhibits highly specific overexpression in cardiac fibroblasts, while showing extremely low expression in cardiomyocytes, cardiac endothelial cells, and cardiac inflammatory cells. Consequently, it is designated as cardiac fibrosis-associated piRNA (CFRPi). In vitro, CFRPi expression was significantly upregulated following fibrosis induction by transforming growth factor-β (TGF-β) or Ang II stimulation, indicating its close association with the fibrotic process. Mechanistically, CFRPi directly targets and binds the 3’UTR of the protective peptide Apelin (APLN) mRNA, inhibiting its translation. Notably, under pressure or injury stimuli, APLN is secreted as a protective peptide, inducing nitric oxide-mediated vasodilation, reducing cardiac workload, and enhancing contractility[47]. Furthermore, APLN expression compensatorily increases in early HF but significantly decreases in advanced stages - exactly opposite to CFRPi’s expression trend. By inhibiting APLN, CFRPi exhibits an inverse expression pattern to APLN during HF progression: lower levels in early stages (2-4 weeks) and significantly elevated levels in advanced stages (8 weeks). This direct interaction was confirmed via dual luciferase assays and further validated in cellular and animal models: CFRPi knockdown significantly increased APLN expression, while its overexpression suppressed APLN. Functionally, CFRPi activates the downstream pro-fibrotic Phosphoinositide 3-kinase/Protein Kinase B/Mammalian Target of Rapamycin (PI3K-AKT-mTOR) signaling pathway by inhibiting APLN. This promotes fibroblast proliferation, myofibroblast differentiation, and collagen secretion in vitro, while exacerbating cardiac fibrosis and worsening cardiac function in vivo. In summary, CFRPi promotes cardiac fibrosis by suppressing the HF protective target APLN and its downstream PI3K/AKT signaling. The CFRPi-APLN-PI3K/AKT signaling axis plays a key regulatory role in the progression of cardiac fibrosis and HF[48] [Figure 2A].
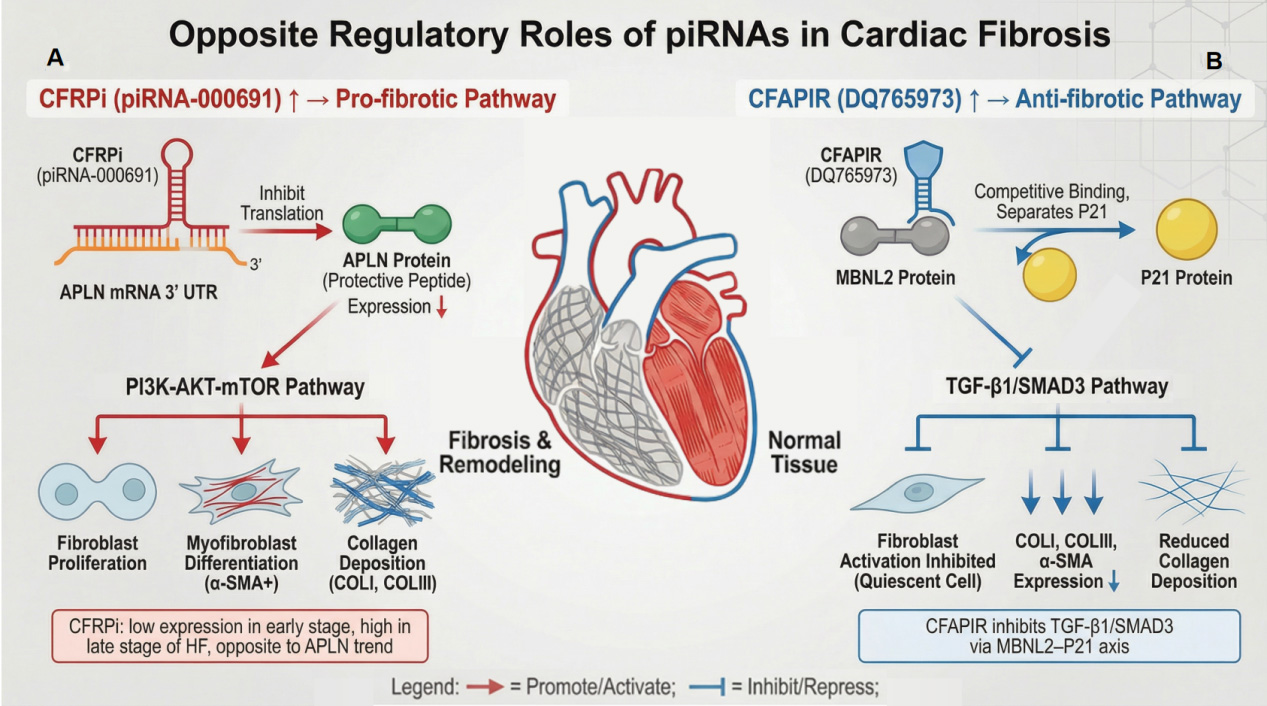
Figure 2. Opposing regulatory roles of piRNAs in cardiac fibrosis. (A) CFRPI-mediated pro-fibrotic pathway. The piRNA CFRPI (piRNA-000691) targets the 3’UTR of APLN mRNA, inhibiting the translation of the protective peptide APLN. Reduced APLN protein levels lead to activation of the PI3K-AKT-mTOR signaling pathway. This cascade promotes fibroblast proliferation, their differentiation into α-SMA-positive myofibroblasts, and subsequent increased deposition of collagen types I and III (COL I, COL III), driving cardiac fibrosis progression; (B) CFAPIR-mediated anti-fibrotic pathway. The piRNA CFAPIR (DQ765973) exerts an anti-fibrotic effect by competitively binding to the MBNL2 protein. This interaction sequesters MBNL2, thereby releasing the bound P21 protein. Elevated P21 activity suppresses the TGF-β1/SMAD3 signaling pathway. Consequently, fibroblast activation is inhibited, maintaining cells in a quiescent state, which leads to reduced expression of α-SMA, COL I, and COL III, and ultimately decreases collagen deposition. Trend Note: CFRPI expression is low in early-stage HF but increases in late stages, showing an opposite trend to APLN expression. HF: Heart failure; APLN: Apelin; piRNA: piwi-interacting RNAs; PI3K-AKT-mTOR: phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin; α-SMA: alpha-smooth muscle actin; TGF-β1: transforming growth factor-β1; 3’UTR: 3’untranslated region; CFAPIR: cardiacfibrosis associated piRNA; CFRPI: cardiacfibrosis associatedpiRNA; SMAD3: mothers against decapentaplegic homolog 3.
Lv et al. identified 20 differentially expressed piRNAs in AngII-treated cardiac fibroblasts via piRNA-chip analysis, among which PiRNA-DQ765973 [designated as cardiac fibrosis-associated piRNA (CFAPIR)] exhibited the most significant downregulation[49]. Moreover, its expression level in cardiac fibroblasts was significantly higher than in cardiomyocytes, macrophages, and endothelial cells. Contrary to CFRPi. However, CFAPIR overexpression markedly improved AngII-induced cardiac dysfunction in rats, manifested as increased left ventricular ejection fraction and short-axis fractional shortening, while also reducing myocardial interstitial fibrosis and expression of related fibrosis markers. Further experiments demonstrated that CFAPIR overexpression effectively suppressed fibroblast activation, proliferation capacity, and soluble collagen secretion while downregulating Collagen I (COL I), COL III, and Alpha-Smooth Muscle Actin (α-SMA) expression, confirming CFAPIR’s direct inhibition of fibroblast-to-myofibroblast transdifferentiation. Experiments revealed that CFAPIR competitively binds Muscleblind-like protein 2 (MBNL2), reducing the interaction between Cyclin-dependent kinase inhibitor 1A (p21) and MBNL2. This subsequently inhibits the activation of the TGF-β1/Mothers against decapentaplegic homolog 3 (SMAD3) signaling pathway, ultimately blocking myocardial fibroblast activation and excessive collagen deposition. Cross-validation experiments further supported this mechanism: co-transfection of CFAPIR and MBNL2 overexpression plasmids in cardiac fibroblasts revealed that MBNL2 overexpression reversed CFAPIR’s suppression of α-SMA, COL I and COL III expression, indicating that CFAPIR’s anti-fibrotic function depends on its regulation of MBNL2[49]. Therefore, the CFAPIR-MBNL2-P21-TGF-β1/SMAD3 signaling axis constitutes an intrinsic anti-fibrotic defense system in the heart [Figure 2B].
tsRNA
tRF-Glu-CTC-013
Li et al. discovered that tRF-Glu-CTC-013 [is tRNA-derived fragment originating from a glutamic acid (Glu) tRNA that possesses the anticodon sequence CTC (where CTC in DNA-based notation corresponds to CUC in the RNA anticodon), with the unique identifier 013] expression is upregulated in an Ang II-induced myocardial hypertrophy model and is highly conserved across species[50]. Transfection with tRF-Glu-CTC-013 Mimic significantly suppressed Ang II-induced increases in cardiomyocyte surface area and downregulated mRNA expression of hypertrophy marker genes Natriuretic Peptide Precursor A (Nppa), Natriuretic Peptide Precursor B (Nppb), and Myh7, indicating direct inhibitory effects of tRF-Glu-CTC-013 on cardiomyocyte hypertrophy.
Further studies revealed that tRF-Glu-CTC-013 also participates in regulating inflammatory and fibrotic pathological processes during myocardial hypertrophy. Regarding inflammation, tRF-Glu-CTC-013 Mimic significantly reduced mRNA expression of inflammatory mediators such as TNF, IL-1β, and IL-6 induced by Ang II. Regarding fibrosis, this tsRNA inhibits the expression of key molecules in the TGF-β/Mothers against decapentaplegic homolog (SMAD) signaling pathway, significantly downregulating mRNA levels of Tgfβ1, Col1a1, Collagen Type III Alpha 1 Chain (Col3a1), and Fibronectin (Fn1), while reducing collagen deposition. Mechanistically, tRF-Glu-CTC-013 directly binds to the 3’UTR region of Taste Receptor Type 1 Member 3 (TAS1R3), suppressing its mRNA and protein expression. Western blot analysis revealed that tRF-Glu-CTC-013 Mimic markedly decreased mTOR phosphorylation and increased the LC3B-II/LC3B-I ratio, indicating enhanced autophagy activity. Concurrently, Tas1r3 interference mimicked this effect, confirming that tRF-Glu-CTC-013 regulates autophagy through the Taste 1 Receptor Member 3 (TASR3)-mTOR axis[50].
In summary, tRF-Glu-CTC-013 serves as a novel biomarker and key regulator of myocardial hypertrophy. By directly binding to and inhibiting TAS1R3 expression, it downregulates mTOR phosphorylation and enhances cardiomyocyte autophagy, thereby exerting antihypertrophic, anti-inflammatory, and antifibrotic protective effects. This study not only deepens our understanding of the pathological mechanisms of myocardial hypertrophy but also provides novel molecular targets for its clinical diagnosis and treatment [Figure 3A].
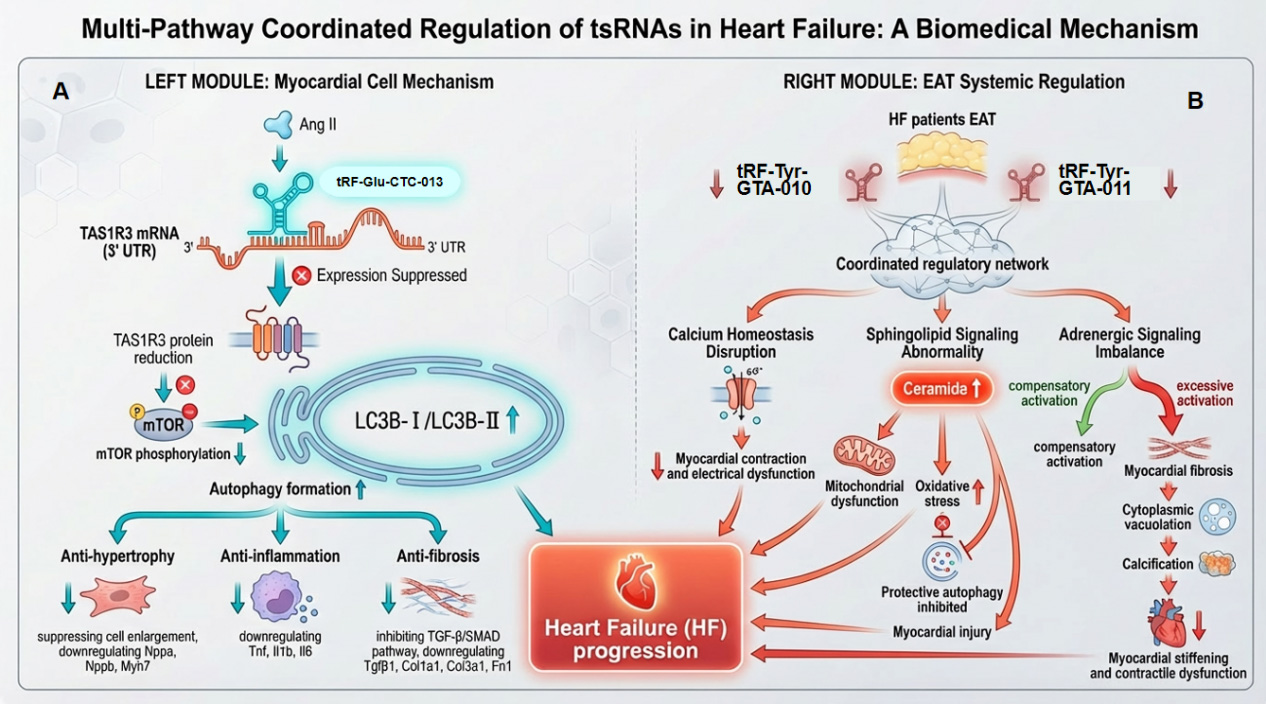
Figure 3. Multi-pathway coordinated regulation of tsRNAs in HF. (A) Myocardial cell-intrinsic protective mechanism. The tsRNA tRF-Glu-CTC-013 binds to the 3’UTR of TAS1R3 mRNA, suppressing TAS1R3 protein expression. Reduced TAS1R3 leads to mTOR phosphorylation inhibition and promotes the conversion of LC3B-I to LC3B-II, thereby inducing autophagy. This autophagic response exerts anti-hypertrophic effects (suppressing cell enlargement and downregulating Nppa, Nppb, Myh7), anti-inflammatory effects (downregulating Tnf, Il1b, Il6), and inhibits the TGF-β/SMAD pathway (downregulating Tgfb1, Col1a1, Col3a1, Fn1). Collectively, this pathway counteracts myocardial hypertrophy, fibrosis, metabolic dysfunction, and subsequent contractile and electrical dysfunction; (B) Systemic regulatory mechanism via epicardial adipose tissue (EAT). In the EAT of HF patients, tsRNAs (IRF-Tyr-GTA-010, IRF-Tyr-GTA-011) form a coordinated regulatory network. This network induces sphingolipid signaling abnormality (increased ceramide) and adrenergic signaling imbalance. These disturbances lead to MF, cytoplasmic vacuolation (fatty degeneration), and calcification, ultimately contributing to myocardial stiffening and contractile dysfunction. tsRNAs: tRNA-derived small RNAs; HF: heart failure; mTOR: mammalian target of rapamycin; TGF-β: transforming growth factor-β; MF: myocardial fibrosis; SMAD: mothers against decapentaplegic homolog.
tRF-Tyr-GTA-010/011
Through RNA sequencing and qRT-PCR validation, this study confirmed the expression patterns of tRF-Tyr-GTA-010 [is tRNA-derived fragment originating from a tyrosine (Tyr) tRNA that possesses the anticodon sequence GTA (where GTA in DNA-based notation corresponds to GUA in the RNA anticodon), with the unique identifier 010] and tRF-Tyr-GTA-011: exhibit a significant downregulation trend in the epicardial adipose tissue (EAT) of HF patients. Bioinformatics analysis further revealed dense interactions among their target genes, suggesting they may form a “joint regulatory effect” by synergistically regulating common target genes, thereby influencing downstream pathophysiological processes[15]. GO enrichment analysis revealed that target genes of tRF-Tyr-GTA-010 and tRF-Tyr-GTA-011 primarily concentrate on regulating calcium ion transport, thereby maintaining normal myocardial contraction and electrophysiological function[51,52]. KEGG pathway analysis revealed that the sphingolipid signaling pathway ranked among the top three pathways enriched for target genes of these two tRFs. Abnormal activation of this pathway is directly associated with the pathological progression of HF[15]. Ceramide, a key sphingolipid molecule, exhibits elevated levels in the circulation and cardiac tissue of HF patients. Elevated ceramide impairs cardiac function through multiple mechanisms: inducing mitochondrial dysfunction to reduce Adenosine Triphosphate (ATP) production, exacerbating oxidative stress to damage myocardial structure, and suppressing protective autophagy to cause accumulation of damaged components[53-57]. Under normal physiological conditions, these two tRFs maintain sphingolipid metabolic balance by suppressing the overexpression of key genes in the sphingolipid pathway. However, their downregulation during HF abnormally activates the sphingolipid pathway, thereby inducing myocardial injury. The adrenergic signaling pathway is another core pathway enriched with target genes of both tRFs. Under physiological conditions, this pathway activates during early HF to enhance myocardial contractility by elevating catecholamine levels, compensating for declining cardiac function. However, prolonged excessive activation induces toxic effects, triggering MF, cytoplasmic vacuolation, and calcification, ultimately leading to myocardial stiffening and further deterioration of contractile function[58-60]. Therefore, tRF-Tyr-GTA-010 and tRF-Tyr-GTA-011 precisely modulate adrenergic signaling pathway activity, ensuring necessary compensatory function while preventing myocardial remodeling damage caused by excessive activation. In HF conditions, downregulation of these two tRFs weakens their regulatory influence on the adrenergic signaling pathway, exacerbating pathway imbalance and worsening cardiac function[15]. In summary, the downregulation of tRF-Tyr-GTA-010 and tRF-Tyr-GTA-011 in HF may jointly promote HF progression by affecting multiple critical pathways, including calcium homeostasis, sphingolipid metabolism, and adrenergic signaling. Restoring their expression holds promise as a novel therapeutic strategy to correct these pathway abnormalities [Figure 3B].
Other hypertrophy-promoting tRFs
tRFs are enriched in hypertrophic hearts, and overexpression of tRFs1 and tRFs2 in H9c2 cells increases cardiomyocyte surface area and expression of cardiac hypertrophy biomarkers[61]. Previous studies also indicate that tsRNAs participate in regulating cardiomyocyte autophagy. In cardiac progenitor cells treated with high glucose, tRF-5014a was found to negatively regulate the expression of the autophagy-related protein Autophagy Related 5 (ATG5). Inhibition of tRF-5014a reversed the suppression of autophagy, enhanced cellular viability, and reduced the release of proinflammatory cytokines under high-glucose conditions[62].
snoRNA
Activation mechanism of the pro-fibrotic snoRNA network
TGF-β can specifically induce a “pro-fibrotic network” composed of 12 snoRNAs in human cardiac fibroblasts, with SNORD42A and SNORA20 showing the most significant upregulation (1.8-2.5-fold). This network exhibits a significant positive correlation with the expression of key extracellular matrix (ECM) synthesis genes (e.g., COL1A1), synergistically amplifying TGF-β signaling to form a positive feedback loop driving cardiac fibrosis. It represents a critical molecular basis for myocardial remodeling in HF. Meanwhile, the sustained overexpression of SNORD113-6, through its unique tRNA modification mechanism, facilitates fibroblast migration and infiltration within the myocardial interstitium. Together, these mechanisms constitute a crucial molecular pathway for the transition from compensatory remodeling to pathological remodeling in MI. Future studies should further analyze the differential expression profiles of distinct snoRNAs across different stages of HF (e.g., acute decompensated phase vs. stable phase) and explore their potential as novel circulating biomarkers for assessing fibrosis burden and prognosis in HF[41,63].
Protective mechanisms of anti-fibrotic snoRNAs
In the chronic progression of HF, sustained imbalance between pro-fibrotic and anti-fibrotic signaling drives progressive myocardial remodeling. Endogenous anti-fibrotic snoRNAs, such as SNORD32A and SNORD51, exhibit persistent downregulation in chronic pathological states, with their expression levels showing significant negative correlation with myocardial collagen content. This chronic downregulation progressively diminishes their translational suppression of the key pro-fibrotic factor peroxisome proliferator-activated receptor delta (Pxdn). Mechanistically, SNORD32A and SNORD51 guide Fibrillarin to methylate Pxdn mRNA, thereby precisely inhibiting its protein synthesis at the translational level. In CHF, the prolonged absence of these protective snoRNAs allows sustained accumulation of Pxdn protein (upregulated 2-3-fold), continuously driving oxidative stress and collagen deposition. Together, these processes contribute to a gradual and irreversible increase in myocardial stiffness[64]. Restoring or enhancing the function of endogenous anti-fibrotic snoRNAs - via mimetics or gene therapy - presents a compelling therapeutic direction for anti-remodeling interventions in HF. Elucidating their cell-specific actions in cardiomyocytes versus non-cardiomyocytes will further inform precision targeting strategies.
Therapeutic potential and clinical prospects of snoRNAs
Building on snoRNA regulatory mechanisms, researchers have developed innovative biotherapeutic strategies. Human cortical stem cell-derived exosomes (hCBSC-dEXOs) naturally enrich multiple anti-fibrotic snoRNAs, effectively reversing fibrotic phenotypes and improving cardiac function in both cellular and animal models - an effect confirmed to depend on their snoRNA content[63,65]. Furthermore, engineered exosomes specifically carrying high doses of SNORD32A/SNORD51 achieve targeted delivery and efficient expression in cardiomyocytes. This significantly suppresses the pro-fibrotic target Pxdn and promotes tissue repair, demonstrating excellent therapeutic potential. SnoRNA therapy represents a paradigm shift in HF treatment from protein regulation to RNA regulation. Future core challenges and opportunities include: developing more efficient and safe targeted delivery systems; exploring snoRNA combination therapies (such as targeting multiple pro-fibrotic and anti-fibrotic members); advancing personalized precision medicine based on snoRNA, ultimately translating these novel ncRNA molecules from basic research to clinical application[41].
DISCUSSION
This review systematically elucidates the pivotal roles of emerging ncRNAs - including piRNAs, tRNAs, and snoRNAs - in the pathogenesis and progression of HF. These findings not only significantly expand our understanding of the complexity of molecular regulatory networks in HF but also challenge the traditional research paradigm that focused solely on miRNAs, lncRNAs, and circRNAs. They reveal a previously overlooked layer of precise regulation by ncRNAs in the pathophysiological process of HF.
Multidimensional mechanisms: complex regulatory networks of non-mainstream ncRNAs in HF
piRNAs, tsRNAs, and snoRNAs - previously regarded as performing only housekeeping functions - exhibit multi-tiered, multi-mechanistic regulatory properties in the context of HF. The actions of these ncRNAs demonstrate distinct cellular and disease-stage specificity. For instance, CHAPIR and HNEAP primarily regulate cardiomyocyte hypertrophy and death, while CFRPi and CFAPIR specifically modulate cardiac fibroblast activation[28,44]. During disease progression, HAAPIR and HNEAP respond to acute injury, whereas CHAPIR, specific tsRNAs, and pro-fibrotic snoRNAs drive chronic remodeling[15,30]. This specificity establishes a theoretical foundation for developing highly selective therapies. These molecules exert effects through highly diverse mechanisms. piRNAs (e.g., CHAPIR) recruit epigenetic modifiers for transcriptional/post-transcriptional regulation; tsRNAs (e.g., tRF-Glu-CTC-013) often mimic miRNA functions by directly targeting mRNAs or modulating their stability; snoRNAs (e.g., SNORD113-6) primarily guide chemical modifications of rRNAs, tRNAs, etc., affecting their function[27,41,50]. They precisely regulate nearly all core pathological processes of HF across multiple levels, including epigenetics, transcription, post-transcriptional processes, and intercellular communication. Studies have revealed functionally antagonistic ncRNA pairs, such as the pro-fibrotic CFRPi and anti-fibrotic CFAPIR, as well as tiRNA promoting apoptosis and counteracting excessive autophagy. The onset and progression of HF may result from disruptions in such endogenous equilibria, suggesting that restoring balance holds greater therapeutic potential than blocking single pathways[48,49].
Significant clinical translation potential: from biomarkers to innovative therapies
These ncRNAs demonstrate remarkable translational promise. Regarding biomarkers, serum piRNA and tsRNA profiles exhibit high stability, showing potential for early diagnosis, risk stratification, and prediction of drug response in AMI. They hold promise as next-generation non-invasive liquid biopsy tools[30]. For therapeutic interventions, studies on gain-of-function (e.g., piRNA-6426, CFAPIR) and loss-of-function (e.g., HAAPIR, HNEAP, CFRPi) provide evidence for two major strategies: “gene supplementation” and “gene silencing”. More innovatively, engineered exosome delivery of snoRNAs (e.g., SNORD32A/SNORD51) has successfully reversed fibrosis in animal models, demonstrating novel targeted therapeutic pathways integrating ncRNAs with exosomes[41,63].
Current challenges and future directions
Despite the field’s promising prospects, several critical challenges remain that demand in-depth research and breakthroughs. First, species-specific differences in preclinical studies pose significant translational barriers. The vast majority of current mechanistic evidence originates from rodent models; however, their ncRNA expression profiles, regulatory networks, and physiological functions exhibit inherent differences from humans. For example, piRNA sequences show low conservation between mice and humans. Although the piwi like RNA-mediated gene silencing 1 (PIWIL) protein and piRNA cluster locations are relatively conserved, low sequence homology limits the extrapolation of functional findings to human HF research[66,67]. Therefore, future efforts should strengthen validation using human cells, organoids, and humanized models, while systematically comparing the conservation and functional differences of ncRNAs across species to enhance the predictive value of preclinical discoveries. Secondly, a core limitation in mechanism understanding lies in incomplete mapping of target networks. Current research predominantly focuses on linear relationships between individual ncRNAs and their direct targets, whereas these molecules actually form intricate interaction networks in vivo. Systematic elucidation remains lacking for their global target profiles, species-specific regulatory circuits across distinct cardiac cell types, and crosstalk with conventional mainstream ncRNAs (e.g., miRNAs, lncRNAs). For example, snoRNAs regulate rRNAs and mRNAs through modifications such as 2’-O-methylation, but their specific RNA targets and functional impacts in HF remain unclear[65]. Furthermore, clinical translational research lags significantly, with very few ongoing clinical trials targeting these ncRNAs as intervention points or core biomarkers. Existing knowledge heavily relies on basic research and observational clinical correlation data, urgently requiring prospective, interventional clinical trials to provide high-level evidence. This includes validating their clinical utility as diagnostic or prognostic biomarkers, as well as evaluating the safety, tolerability, and preliminary efficacy of strategies based on silencing (e.g., ASO, siRNA) or supplementation (e.g., mimetics, gene therapy). Initiating and rigorously designing such clinical trials represents a decisive step in evaluating their ultimate translational value[68]. Finally, this field also holds significant opportunities to advance personalized medicine. Preliminary studies indicate that the expression profiles of ncRNAs such as tsRNA correlate with drug response heterogeneity, suggesting these molecules may guide personalized HF treatment in the future. Further exploration of their associations with patient genetic backgrounds, etiological subtypes, and clinical phenotypes will be a key pathway toward achieving precision medicine for HF.
CONCLUSION
HF, as a complex clinical syndrome, is closely associated with the imbalance of molecular regulatory networks in its onset and progression. Traditionally, research has primarily focused on the roles of miRNAs, lncRNAs, and circRNAs in maintaining cardiac homeostasis and pathological remodeling. However, this review systematically reveals that ncRNAs such as piRNAs, tRNAs, and snoRNAs play indispensable and increasingly well-defined roles in HF, particularly during its acute decompensated and chronic progressive stages [Table 1].
Summary of functions and characteristics of non-coding RNAs
| RNA | Cell source | Disease stage | Molecular targets | Functional roles |
| HAAPIR[27] | Cardiomyocytes | AHF | NAT10 | HAAPIR-NAT10-TFEC-BIK signaling axis |
| HNEAP[28] | Cardiomyocytes | AHF | DNMT1 | Competitive binding to MBNL2 reduces interactions between P21 and MBNL2, thereby inhibiting the TGF-β1/SMAD3 signaling pathway and alleviating fibrosis |
| tiRNA-HAR (AS-tDR-001449)[32] | Cardiomyocytes | AHF | HuR, p53 mRNA | HuR binds to and enhances its interaction with p53 mRNA, stabilizing p53 mRNA, upregulating p53 protein, promoting cardiomyocyte apoptosis, and worsening cardiac function |
| tiRNA-Met-CAT-002[34] | Cardiomyocytes | AHF | The 3’UTR region of Bnip3 mRNA | By targeting the 3’UTR of Bnip3 mRNA, this approach suppresses Bnip3 expression, reduces the LC3B-II/LC3B-I ratio and Beclin-1 expression, inhibits excessive autophagy in cardiomyocytes, and mitigates I/R injury |
| SNORD113-6[40] | Cardiomyocytes, coronary vascular cells | AHF | The anticodon region of specifically methylated tRNALeu | SNORD113-6 maintains tRNA stability through methylation, suppresses abnormal tRF generation, and protects myocardial metabolism and vascular homeostasis |
| CFRPi (piRNA-000691)[48] | Cardiac fibroblasts | CHF | The 3’UTR region of APLN mRNA | Inhibits APLN mRNA translation, activates the PI3K-AKT-mTOR pathway, and exacerbates cardiac fibrosis |
| CFAPIR (DQ765973)[49] | Cardiac fibroblasts | CHF | MBNL2 | Competitively binds MBNL2, inhibits TGF-β1/SMAD3 signaling pathway, and alleviates fibrosis |
| tRF-Glu-CTC-013[50] | Cardiomyocytes | CHF | The 3’UTR region of TAS1R3 mRNA | Inhibits TAS1R3 expression, enhances cardiomyocyte autophagy, and exerts antihypertrophic, anti-inflammatory, and antifibrotic effects |
| tRF-Tyr-GTA-010/011[15] | Visceral adipose tissue in heart failure patients | CHF | Calcium transport-related genes, Sphingolipid signaling pathway genes, Adrenergic signaling pathway genes | Downregulation of expression leads to calcium dysregulation, excessive activation of the sphingolipid pathway and adrenergic signaling, accelerating myocardial remodeling and deterioration of cardiac function |
| SNORD32A/SNORD51[64] | Cardiac fibroblasts, cardiomyocytes | CHF | Pxdn mRNA | Methylation of Pxdn mRNA inhibits its translation, reduces oxidative stress and collagen deposition, and delays fibrosis |
In AHF, these molecules rapidly respond to stress signals such as ischemia and hypoxia, becoming key decision-makers in cellular fate. For example, piRNA-HAAPIR regulates ac4C modification or m5C methylation activity, forming the HAAPIR-NAT10-TFEC-BIK signaling axis that drives CA. piRNA-HNEAP interacts with DNMT1, inhibiting its RNA m5C methylation activity, leading to upregulation of ATF7 expression and thereby driving necroptosis[28]; tiRNAs (e.g., tiRNA-HAR, tiRNA-Met-CAT-002), regulated by the HIF1α/ANG axis, mitigate excessive autophagy in cardiomyocytes and enhance cell viability by affecting p53 stability or binding to the 3’UTR of Bnip3 to suppress its expression. This ultimately exerts I/R injury protection, precisely regulating cellular homeostasis[34]. Concurrently, disrupted expression profiles of serum piRNAs and tsRNAs demonstrate significant potential as non-invasive biomarkers for early diagnosis, risk stratification, and treatment response prediction in AMI[29]. snoRNAs (such as members of the 14q32 clusters) mediate chemical modifications of RNAs such as tRNAs, coordinating vascular remodeling and myocardial metabolic adaptation during acute stress. Their intercellular communication via EVs further amplifies injury signals[41].
In CHF, these molecules are deeply involved in the core process of pathological myocardial remodeling, exhibiting precise cell-specific regulation and functional antagonism. piRNAs such as CHAPIR drive myocardial hypertrophy by interfering with m6A methylation, while CFRPi and CFAPIR exert opposing pro-fibrotic and anti-fibrotic effects in cardiac fibroblasts by inhibiting APLN and competitively binding MBNL2, respectively, forming a complex regulatory network[44]. tsRNAs (e.g., tRF-Glu-CTC-013, tRF-Tyr-GTA-010/011) exert multifaceted protective effects by targeting TAS1R3, calcium homeostasis, and neuroendocrine pathways, thereby suppressing hypertrophy, inflammation, and fibrosis. snoRNAs form distinct pro-fibrotic (e.g., TGF-β-induced SNORD42A network) and anti-fibrotic (e.g., SNORD32A/SNORD51) factions, whose sustained functional imbalance drives irreversible myocardial remodeling[50]. Notably, therapeutic strategies based on snoRNAs (e.g., delivering SNORD32A/SNORD51 via engineered exosomes) have demonstrated significant effects in preclinical models by reversing fibrosis and improving cardiac function. This marks a paradigm shift in HF treatment, moving from targeting proteins to precisely regulating RNA molecules[41,63].
Of particular note is that despite demonstrating potential therapeutic value in preclinical models, therapies based on molecules such as piRNAs still face multiple critical challenges in translating to clinical applications: (1) Complex biological properties and mechanisms of action: piRNA stability is influenced by terminal modifications, sequence characteristics, length, and the type of P-element induced wimpy testis (PIWI) protein it binds to, with significant interspecies variations. This makes it difficult to directly extrapolate findings from animal models to humans[69]. Even though piRNAs were observed to cleave hundreds of transcripts in mouse models, they significantly altered the steady-state expression levels of only a few targets, suggesting that piRNA action is highly selective and complex, posing challenges for predicting and regulating therapeutic effects[70]. Furthermore, the biogenesis and silencing mechanisms of piRNAs are more complex than those of miRNAs/siRNAs, further limiting the rational design and optimization of related therapeutic strategies[71]; (2) Limitations in Delivery Efficiency and Bioavailability: Although therapeutic effects have been observed in xenograft models and organoids, piRNA delivery efficiency, tissue targeting, and bioavailability in humans require substantial improvement - a prerequisite for clinical application[72]; (3) Safety and Off-Target Effects Risk: piRNAs may induce off-target effects through incomplete complementary pairing, disrupting the expression of unintended genes and potentially triggering toxic reactions[73]. The piRNA-PIWI complex plays a central role in regulating transposon silencing and maintaining genomic stability; long-term treatment may introduce uncertain risks at the genetic level[74]. Furthermore, the critical role of piRNAs in germ cells raises concerns about reproductive toxicity, limiting their applicability in certain disease areas[75]; (4) Translational limitations of preclinical models: piRNA expression and function exhibit marked gender and tissue specificity. Effects observed in animal models may not accurately reflect real-world responses across different human genders, tissues, or pathological states[76]; (5) Regulatory and ethical considerations: piRNA therapies involve modulating genomic stability and epigenetic states, raising ethical concerns about such interventions that require careful balancing of risks and benefits[77]. Currently, specific regulatory standards for novel RNA therapies such as piRNA remain underdeveloped, with significant uncertainties persisting in clinical trial design, efficacy assessment, and safety monitoring[78].
In summary, in-depth exploration of the roles of piRNAs, tRNAs, and snoRNAs in HF has greatly enriched our understanding of the molecular regulatory landscape of this disease. These molecules not only serve as key to unlocking novel pathophysiological mechanisms of HF but also emerge as potential novel biomarkers and therapeutic targets, opening promising new avenues for developing innovative precision diagnostics and personalized treatment strategies for HF. Future research will continue to bridge knowledge gaps, propelling these RNAs from basic science to clinical practice, ultimately contributing to overcoming HF as a global health challenge.
DECLARATIONS
Authors’ contributions
Conceptualization, methodology, writing - original draft, visualization: Wang X, Li B, Gao J, Ma Z
Writing - review and editing, reviewed and made significant revisions to the manuscript: Yang S, Zhang M, Wang K
All authors have read and approved the final manuscript. All authors provided direction and guidance throughout the preparation of this manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Lovart (Official version, released 2025-7-23) was used to create Figures 2, 3, and the graphical abstract (GA). The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (82370291); the Open Project Program of the State Key Laboratory of Frigid Zone Cardiovascular Diseases (SKLFZCD), Harbin Medical University (HDHY2024007); the Qingdao Science and Technology Benefiting the People Demonstration Project (24-1-8-smjk-7-nsh); the Major Basic Research Projects in Shandong Province (ZR2024ZD46); and the Taishan Scholar Distinguished Expert Program.
Conflicts of interest
Wang K. is an Editorial Board Member of the journal Vessel Plus. Wang K. was not involved in any steps of the editorial process, including reviewer selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Lippi G, Sanchis-gomar F. Global epidemiology and future trends of heart failure. AME Med J. 2020;5:15.
4. Pastore MC, Vigna M, Saglietto A, et al. Prognostic value of left atrial strain in acute and chronic heart failure: a meta-analysis. ESC Heart Fail. 2025;12:2921-31.
5. Shah KS, Xu H, Matsouaka RA, et al. Heart failure with preserved, borderline, and reduced ejection fraction: 5-year outcomes. J Am Coll Cardiol. 2017;70:2476-86.
6. Vilella-Figuerola A, Gallinat A, Escate R, Mirabet S, Padró T, Badimon L. Systems biology in chronic heart failure-identification of potential miRNA regulators. Int J Mol Sci. 2022;23.
7. Gheorghiade M, Zannad F, Sopko G, et al. ; International Working Group on Acute Heart Failure Syndromes. Acute heart failure syndromes: current state and framework for future research. Circulation. 2005;112:3958-68.
8. Deniau B, Costanzo MR, Sliwa K, Asakage A, Mullens W, Mebazaa A. Acute heart failure: current pharmacological treatment and perspectives. Eur Heart J. 2023;44:4634-49.
9. Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20:5-20.
11. Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD. PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet. 2019;20:89-108.
12. Aravin AA, Sachidanandam R, Bourc'his D, et al. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 2008;31:785-99.
13. Gu X, Zhang Y, Qin X, Ma S, Huang Y, Ju S. Transfer RNA-derived small RNA: an emerging small non-coding RNA with key roles in cancer. Exp Hematol Oncol. 2022;11:35.
14. Schaefer M, Pollex T, Hanna K, et al. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010;24:1590-5.
15. Zhao L, Peng Y, Su P. Expression profiles and functional analysis of tRNA-derived small RNAs in epicardial adipose tissue of patients with heart failure. Ann Med. 2023;55:2267981.
16. Su Z, Wilson B, Kumar P, Dutta A. Noncanonical roles of tRNAs: tRNA fragments and beyond. Annu Rev Genet. 2020;54:47-69.
17. Cao J, Cowan DB, Wang DZ. tRNA-derived small RNAs and their potential roles in cardiac hypertrophy. Front Pharmacol. 2020;11:572941.
18. Zhang L, Liu J, Hou Y. Classification, function, and advances in tsRNA in non-neoplastic diseases. Cell Death Dis. 2023;14:748.
19. Kiss T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001;20:3617-22.
20. Kishore S, Stamm S. Regulation of alternative splicing by snoRNAs. Cold Spring Harbor Symp Quant Biol. 2006;71:329-34.
21. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;79:e263-421.
22. Mann DL, Felker GM. Mechanisms and models in heart failure: a translational approach. Circ Res. 2021;128:1435-50.
23. Du B, Fu Q, Yang Q, et al. Different types of cell death and their interactions in myocardial ischemia-reperfusion injury. Cell Death Discov. 2025;11:87.
24. McDonagh TA, Metra M, Adamo M, et al. ; ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42:3599-726.
25. Yang J, Xue FT, Li YY, Liu W, Zhang S. Exosomal piRNA sequencing reveals differences between heart failure and healthy patients. Eur Rev Med Pharmacol Sci. 2018;22:7952-61.
26. Zhong N, Nong X, Diao J, Yang G. piRNA-6426 increases DNMT3B-mediated SOAT1 methylation and improves heart failure. Aging. 2022;14:2678-94.
27. Wang K, Zhou LY, Liu F, et al. PIWI-interacting RNA HAAPIR regulates cardiomyocyte death after myocardial infarction by promoting NAT10-mediated ac4C acetylation of Tfec mRNA. Adv Sci. 2022;9:e2106058.
28. Wang K, Li FH, Zhou LY, et al. HNEAP regulates necroptosis of cardiomyocytes by suppressing the m5C Methylation of Atf7 mRNA. Adv Sci. 2023;10:e2304329.
29. Huang Y, Li Y, Zhang K, et al. Expression and diagnostic value of PIWI-interacting RNA by serum in acute myocardial infarction. J Cardiol. 2023;82:441-7.
30. Law PT, Qin H, Ching AK, et al. Deep sequencing of small RNA transcriptome reveals novel non-coding RNAs in hepatocellular carcinoma. J Hepatol. 2013;58:1165-73.
31. Guo J, Chen X, Ren J, Wang Y, Wang K, Yang S. The role of tRNA-derived small RNAs (tsRNAs) in regulating cell death of cardiovascular diseases. Biology. 2025;14:218.
32. Wang C, Gao Y, Liu K, et al. tiRNA-HAR contributes to ischemic myocardial injury via facilitating HuR-mediated stability of p53. Transl Res. 2025;280:17-28.
33. Tao EW, Wang HL, Cheng WY, Liu QQ, Chen YX, Gao QY. A specific tRNA half, 5’tiRNA-His-GTG, responds to hypoxia via the HIF1α/ANG axis and promotes colorectal cancer progression by regulating LATS2. J Exp Clin Cancer Res. 2021;40:67.
34. Deng L, Weng Y, Lin J, et al. A tRNA-derived fragment tiRNA-Met-CAT-002 induced by myocardial ischemia/reperfusion injury inhibits cardiomyocyte autophagy by regulating Bnip3. Exp Cell Res. 2025;453:114809.
35. Santovito D, Steffens S, Barachini S, Madonna R. Autophagy, innate immunity, and cardiac disease. Front Cell Dev Biol. 2023;11:1149409.
36. Su J, Cheng J, Hu Y, et al. Transfer RNA-derived small RNAs and their potential roles in the therapeutic heterogeneity of sacubitril/valsartan in heart failure patients after acute myocardial infarction. Front Cardiovasc Med. 2022;9:961700.
37. Greene SJ, Fonarow GC, DeVore AD, et al. Titration of medical therapy for heart failure with reduced ejection fraction. J Am Coll Cardiol. 2019;73:2365-83.
38. Goossens EAC, Zhang L, de Vries MR, Jukema JW, Quax PHA, Nossent AY. Cold-inducible RNA-binding protein but not its antisense lncRNA is a direct negative regulator of angiogenesis in vitro and in vivo via regulation of the 14q32 angiomiRs-microRNA-329-3p and microRNA-495-3p. Int J Mol Sci. 2021;22:12678.
39. Håkansson KEJ, Goossens EAC, Trompet S, et al. Genetic associations and regulation of expression indicate an independent role for 14q32 snoRNAs in human cardiovascular disease. Cardiovasc Res. 2019;115:1519-32.
40. van Ingen E, Engbers PAM, Woudenberg T, et al. C/D box snoRNA SNORD113-6 guides 2’-O-methylation and protects against site-specific fragmentation of tRNA(Leu)(TAA) in vascular remodeling. Mol Ther Nucleic Acids. 2022;30:162-72.
41. Chabronova A, Holmes TL, Hoang DM, et al. SnoRNAs in cardiovascular development, function, and disease. Trends Mol Med. 2024;30:562-78.
42. Ni Y, Deng J, Bai H, Liu C, Liu X, Wang X. CaMKII inhibitor KN-93 impaired angiogenesis and aggravated cardiac remodelling and heart failure via inhibiting NOX2/mtROS/p-VEGFR2 and STAT3 pathways. J Cell Mol Med. 2022;26:312-25.
43. Li Y, Ruan X, Xu X, et al. Shengmai injection suppresses angiotensin II-induced cardiomyocyte hypertrophy and apoptosis via activation of the AMPK signaling pathway through energy-dependent mechanisms. Front Pharmacol. 2019;10:1095.
44. Gao XQ, Zhang YH, Liu F, et al. The piRNA CHAPIR regulates cardiac hypertrophy by controlling METTL3-dependent N6-methyladenosine methylation of Parp10 mRNA. Nat Cell Biol. 2020;22:1319-31.
45. Alcaide P, Kallikourdis M, Emig R, Prabhu SD. Myocardial Inflammation in Heart Failure With Reduced and Preserved Ejection Fraction. Circ Res. 2024;134:1752-66.
46. Wu X, Wang R, Chen X, Wang K. Advances in RNA modification in myocardial fibrosis (Review). Int J Mol Med. 2025;56:218.
47. Kuba K, Zhang L, Imai Y, et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ Res. 2007;101:e32-42.
48. Chen B, Shi B, Zhou Z, et al. Targeting a cardiac abundant and fibroblasts-specific piRNA (CFRPi) to attenuate and reverse cardiac fibrosis in pressure-overloaded heart failure. Transl Res. 2024;267:10-24.
49. Lv L, Yuan K, Li J, et al. PiRNA CFAPIR inhibits cardiac fibrosis by regulating the muscleblind-like protein MBNL2. Biochim Biophys Acta Mol Basis Dis. 2024;1870:167456.
50. Li W, Lan M, Xu K, et al. Screening and evaluation of tRF-Glu-CTC-013 as a biomarker and key regulator in the development of cardiac hypertrophy. Int J Med Sci. 2025;22:2839-51.
51. Jung P, Seibertz F, Fakuade FE, et al. Increased cytosolic calcium buffering contributes to a cellular arrhythmogenic substrate in iPSC-cardiomyocytes from patients with dilated cardiomyopathy. Basic Res Cardiol. 2022;117:5.
52. Hutchings DC, Madders GWP, Niort BC, et al. Interaction of background Ca2+ influx, sarcoplasmic reticulum threshold and heart failure in determining propensity for Ca2+ waves in sheep heart. J Physiol. 2022;600:2637-50.
53. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709-24.
54. Lemaitre RN, Jensen PN, Hoofnagle A, et al. Plasma ceramides and sphingomyelins in relation to heart failure risk. Circ Heart Fail. 2019;12:e005708.
55. Yu J, Pan W, Shi R, et al. Ceramide is upregulated and associated with mortality in patients with chronic heart failure. Can J Cardiol. 2015;31:357-63.
56. Sasset L, Manzo OL, Zhang Y, et al. Nogo-a reduces ceramide de novo biosynthesis to protect from heart failure. Cardiovasc Res. 2023;119:506-19.
57. RETRACTION: α-Synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 2024;38:e70007. [PMID: 39177105].
58. Santulli G, Iaccarino G. Adrenergic signaling in heart failure and cardiovascular aging. Maturitas. 2016;93:65-72.
59. Lin LY, Wu CK, Juang JM, et al. Myocardial regional interstitial fibrosis is associated with left intra-ventricular dyssynchrony in patients with heart failure: a cardiovascular magnetic resonance study. Sci Rep. 2016;6:20711.
60. Trial J, Entman ML, Cieslik KA. Mesenchymal stem cell-derived inflammatory fibroblasts mediate interstitial fibrosis in the aging heart. J Mol Cell Cardiol. 2016;91:28-34.
61. Shen L, Gan M, Tan Z, et al. A novel class of tRNA-derived small non-coding RNAs respond to myocardial hypertrophy and contribute to intergenerational inheritance. Biomolecules. 2018;8:54.
62. Zhao Y, Wang R, Qin Q, Yu J, Che H, Wang L. Differentially expressed tRNA-derived fragments and their roles in primary cardiomyocytes stimulated by high glucose. Front Endocrinol. 2022;13:1049251.
63. Schena GJ, Murray EK, Hildebrand AN, et al. Cortical bone stem cell-derived exosomes’ therapeutic effect on myocardial ischemia-reperfusion and cardiac remodeling. Am J Physiol Heart Circ Physiol. 2021;321:H1014-29.
64. Elliott BA, Ho HT, Ranganathan SV, et al. Modification of messenger RNA by 2’-O-methylation regulates gene expression in vivo. Nat Commun. 2019;10:3401.
65. Sun X, Wang G, Luo W, et al. Small but strong: the emerging role of small nucleolar RNA in cardiovascular diseases. Front Cell Dev Biol. 2023;11:1292925.
66. Shivam P, Ball D, Cooley A, et al. Regulatory roles of PIWI-interacting RNAs in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2025;328:H991-H1004.
67. Cheang I, Zhu Q, Liao S, Li X. Current understanding of piRNA in cardiovascular diseases. Front Mol Med. 2021;1:791931.
68. Das S, Shah R, Dimmeler S, et al. ; American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology. Noncoding RNAs in cardiovascular disease: current knowledge, tools and technologies for investigation, and future directions: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2020;13:e000062.
69. Gainetdinov I, Colpan C, Cecchini K, et al. Terminal modification, sequence, length, and PIWI-protein identity determine piRNA stability. Mol Cell. 2021;81:4826-4842.e8.
70. Cecchini K, Ajaykumar N, Bagci A, Zamore PD, Gainetdinov I. Mouse pachytene piRNAs cleave hundreds of transcripts, but alter the steady-state abundance of only a minority of targets. bioRxiv. 2024.
71. Czech B, Hannon GJ. One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci. 2016;41:324-37.
72. Qin HL, Han Y, Li JQ, et al. piRNA28846 has the potential to be a novel RNA nucleic acid drug for ovarian cancer. NPJ Precis Oncol. 2025;9:65.
73. Bereczki Z, Benczik B, Balogh OM, et al. Mitigating off-target effects of small RNAs: conventional approaches, network theory and artificial intelligence. Br J Pharmacol. 2025;182:340-79.
74. Bartoszewski R, Sikorski AF. Editorial focus: understanding off-target effects as the key to successful RNAi therapy. Cell Mol Biol Lett. 2019;24:69.
75. Czech B, Munafò M, Ciabrelli F, et al. piRNA-guided genome defense: from biogenesis to silencing. Annu Rev Genet. 2018;52:131-57.
76. Perera BPU, Wang K, Wang D, et al. Sex and tissue-specificity of piRNA regulation in adult mice following perinatal lead (Pb) exposure. Epigenetics. 2024;19:2426952.
77. Li Y, Bhinge A, Inoue S, Garcia G. Editorial: Noncoding RNAs in neurodegenerative disorders: from current insights and future directions to translational modeling and therapeutic approaches. Front Neurosci. 2024;18:1497673.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].