


Integrin signaling pathways in pulmonary hypertension
 0
0 Abstract
Pulmonary hypertension (PH) is a progressive and life-threatening disorder characterized by elevated pulmonary arterial pressure, vascular remodeling, and right ventricular failure. While the pathogenesis of PH involves endothelial dysfunction, inflammation, and excessive extracellular matrix (ECM) deposition, emerging evidence highlights the pivotal role of integrin-mediated signaling in driving vascular cell behavior and tissue stiffness. Integrins, a family of heterodimeric transmembrane receptors, serve as critical mechanosensors and signal transducers between cells and the ECM. The dysregulation of integrins has been confirmed to promote pathological vascular remodeling through the following mechanisms: (1) Activating focal adhesion kinase (FAK) and Src family kinases, driving excessive proliferation and resistance to apoptosis of pulmonary artery smooth muscle cells; (2) Enhanced transforming growth factor-beta (TGF-β) signaling leads to the transformation of fibroblasts into myofibroblasts and excessive collagen deposition; (3) Ras homolog gene family, member A/Rho-associated protein kinase-mediated cytoskeletal recombination disrupts the integrity of the endothelial barrier, exacerbating inflammation and thrombosis. These pathways collectively increase vascular hardness and maintain a pro-remodeling microenvironment of pulmonary vessels. This review summarizes the current understanding of integrin signaling pathways in PH, with a focus on αvβ3, α5β1, and β1-containing integrins, their downstream effectors (e.g., FAK, TGF-β), and their interplay with inflammatory and fibrotic processes. We also discuss preclinical and clinical evidence supporting integrin-targeted therapies, including Myocardin-related transcription factor 1 and Cilengitide, as potential strategies for modulating vascular remodeling in PH. However, their clinical transformation remains challenged by limited efficacy, context-dependent signaling, and safety concerns. A deeper understanding of integrin biology may facilitate the development of more precise and effective therapeutic strategies for PH.
Keywords
INTRODUCTION
Pulmonary hypertension (PH) is a progressive and fatal cardiopulmonary disease characterized by sustained elevation of pulmonary artery pressure. The progression of the disease will gradually lead to right heart failure and even death[1-3]. In clinical diagnosis, a resting mean pulmonary arterial pressure (mPAP)
The core mechanism of PH pathogenesis lies in the pathological remodeling of the pulmonary vascular system, particularly changes in small arteries[9]. This process is not an independent behavior of a single cell type but involves complex dynamic interactions between pulmonary arterial endothelial cells (PAECs), pulmonary arterial smooth muscle cells (PASMCs), fibroblasts, immune cells, and extracellular matrix (ECM) components[10]. It is mainly characterized by abnormal proliferation and apoptosis resistance of PASMCs, leading to medial hypertrophy; endothelial dysfunction-driven intimal hyperplasia and plexiform lesion formation; local coagulation imbalance causing in situ thrombosis; and accompanying perivascular inflammatory cell infiltration and interstitial fibrosis[11-13].
Currently, PH treatment strategies mainly focus on improving the imbalance of soluble vasoactive mediators, such as excessive endothelin-1 (ET-1) production, inhibition of nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) pathways, and decreased prostacyclin (PGI2) synthesis[1,13]. Although these treatments have shown some success in alleviating vasoconstriction, they are still inadequate to reverse structural vascular lesions, suggesting that modulating vascular tone alone is insufficient to effectively halt the progression of PH. In contrast, targeting integrins offers a novel approach that not only addresses vascular tone but also directly modulates the underlying vascular remodeling in PH. This innovative therapeutic strategy has the potential to reverse pathological ECM deposition and restore vascular homeostasis, representing a significant step beyond traditional vasodilator therapies.
Recent studies indicate that dynamic changes of the ECM and the mechanical signal perception system play crucial roles in driving persistent pulmonary vascular remodeling[14,15]. Stimulated by factors such as chronic hypoxia, inflammation, abnormal shear stress, or genetic predisposition, the pulmonary vascular microenvironment undergoes significant changes, manifested as abnormal deposition of ECM components like fibronectin, collagen, and tenascin-C, increased matrix stiffness, and degradation imbalance[16]. These physical and biochemical signals are sensed by cells via specific receptors, especially integrins, thereby activating downstream signaling networks[14,17]
Integrins are key transmembrane receptors that connect cells to the ECM[18,19]. Apart from mediating cell adhesion and anchorage, integrins also act as signaling hubs that regulate cell proliferation, migration, phenotype transformation, and survival[20]. Under the pathological conditions of PH, specific integrin subtypes (e.g., α5β1, αvβ3, αvβ5) are upregulated in PASMCs and PAECs, activating downstream signaling pathways such as focal adhesion kinase (FAK), Src, phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT), and mitogen-activated protein kinase (MAPK), promoting abnormal cell proliferation and apoptosis resistance[21-23]. Moreover, activation of certain integrins (e.g., αvβ6/αvβ8) may activate latent transforming growth factor-beta (TGF-β), thereby amplifying fibrosis and inflammation[24-27]. Integrins also interact with other mechanosensitive pathways that influence pulmonary vascular remodeling—most notably the Hippo/YAP (Yes-associated protein) pathway. In PH, elevated ECM stiffness promotes integrin activation and downstream FAK signaling, which inhibits the Hippo pathway, leading to nuclear translocation of the transcriptional co-activators YAP/TAZ (transcriptional co-activator with PDZ-binding motif) to promote the transcription of genes related to cell proliferation, survival and fibrosis. Critically, YAP/TAZ also feedback to upregulate integrin and matrix components, increasing ECM stiffness and further amplifying integrin signaling. This bidirectional crosstalk establishes a self-sustaining vicious cycle that continuously aggravates pulmonary vascular remodeling in PH[28-30]. Therefore, integrin-mediated “cell-ECM crosstalk" is not only an important regulatory factor in vascular YAP/TAZ structural remodeling but also provides a potential breakthrough for the development of novel therapeutic strategies targeting structural lesions.
This review comprehensively summarizes the latest research on integrin signaling in PH, discusses the dysregulation of specific integrin subtypes in pulmonary vasculature, introduces the main downstream signaling pathways, explores their interactions with growth factors and inflammatory mediators, and evaluates the therapeutic potential of targeting integrins and their effectors. Additionally, we highlight the challenges in translating these insights into clinical practice and future research directions.
INTEGRIN STRUCTURE AND FUNCTION
Integrins are a group of widely expressed, heterodimeric transmembrane glycoprotein receptors on the cell surface, formed by non-covalent bonding between α and β subunits[31]. Since their discovery in 1980s, research has shown that mammals express at least 18 α subunits and 8 β subunits, which combine to form 24 different integrin subtypes[32,33]. Each integrin subunit typically consists of a large extracellular domain for ligand binding, a single transmembrane helix, and a short cytoplasmic tail that links to intracellular signaling and structural proteins[34] [Table 1].
The 24 known integrin heterodimers
| Main ligand | Expressing cells | References | |
| α1β1 (CD49a/CD29) | Collagen I, IV, Laminin | Fibroblasts, smooth muscle cells, some T cells | [35-37] |
| α2β1 (CD49b/CD29) | Collagen I, III, IV | Platelets, epithelial cells, fibroblasts | [38,39] |
| α3β1 (CD49c/CD29) | Laminin, Fibronectin, Thrombospondin | Epithelial, endothelial, fibroblasts | [40-45] |
| α4β1 (VLA-4, CD49d/CD29) | Fibronectin (CS-1), VCAM-1 | Lymphocytes, monocytes, eosinophils | [46-48] |
| α5β1 (VLA-5, CD49e/CD29) | Fibronectin (RGD), Fibrillin, Thrombospondin | Widely expressed (fibroblasts, SMCs, endothelia, blood cells) | [49-54] |
| α6β1 (CD49f/CD29) | Laminin isoforms (laminin-111, laminin-511, etc.) | Epithelial cells, stem cells, platelets | [40,55] |
| α7β1 (CD49g/CD29) | Laminin | Skeletal and cardiac muscle cells | [56-58] |
| α8β1 (CD49h/CD29) | Fibronectin, Vitronectin, Osteopontin | Smooth muscle, neurons, kidney cells | [59-61] |
| α9β1 (CD49i/CD29) | Fibronectin, VCAM-1, Tenascin-C, Osteopontin, ADAMs | Leukocytes, epithelial cells, endothelial cells | [62,63] |
| α10β1 (CD49j/CD29) | Collagen I, II, IV | Chondrocytes, fibroblasts | [64,65] |
| α11β1 (CD49k/CD29) | Collagen I, II, III | Fibroblasts, mesenchymal cells | [38,66] |
| αvβ1 | Fibronectin, Vitronectin, Latent TGF-β | Fibroblasts, smooth muscle cells, epithelial cells | [59,67,68] |
| αLβ2 (CD11a/CD18) | ICAM-1,2,3 | T/B lymphocytes, monocytes | [46,69-71] |
| αMβ2 (CD11b/CD18) | iC3b, ICAM-1, Fibrinogen | Neutrophils, monocytes, macrophages | [46,70,72] |
| αXβ2 (CD11c/CD18) | iC3b, Fibrinogen | Dendritic cells, monocytes | [73-75] |
| αDβ2 (CD11d/CD18) | ICAM-3, VCAM-1 | Monocytes, macrophages | [69,70,75,76] |
| αIIbβ3 (CD41/CD61) | fibrinogen, fibronectin, vWF | Platelets (exclusively) | [59,77] |
| αvβ3 (CD51/CD61) | Vitronectin, Fibronectin, Osteopontin, Tenascin | Endothelial cells, osteoclasts, tumor cells, smooth muscle cells | [59,78-80] |
| α6β4 | Laminin-332 (laminin-5) | Epithelial cells (localized in hemidesmosomes) | [40,81,82] |
| αvβ5 | Vitronectin | Fibroblasts, epithelial, endothelial, tumor cells | [59,83-87] |
| αvβ6 | Fibronectin, Tenascin, Latent TGF-β | Epithelial cells (low in normal tissue, upregulated in injury/tumor) | [59,88-91] |
| α4β7 | MAdCAM-1, Fibronectin | Gut-homing lymphocytes | [92,93] |
| αEβ7 | E-cadherin | Intraepithelial lymphocytes (IELs), regulatory T cells | [94-96] |
| αvβ8 | Latent TGF-β | Neurons, glial cells, smooth muscle cells, epithelial cells | [97,98] |
According to the different ligands identified, integrins are classified into four categories [Table 2][19,103,104]:
Classification of integrins based on ligand specificity
| Integrin category | Representative heterodimers | Primary ligands/binding motifs | Key features and notes | References |
| Collagen-binding integrins | α1β1, α2β1, α10β1, α11β1 | Collagens (GFOGER-like motifs), laminins (for α1β1, α2β1) | Different specificities: α1β1 prefers collagen IV, α2β1 binds collagen I-III, α10β1 in cartilage, α11β1 in mesenchymal cells cell adhesion, migration, and angiogenesis | [38] |
| Laminin-binding integrins | α3β1, α6β1, α7β1, α6β4 | Laminins (basement membrane, multiple isoforms) | α7β1 abundant in skeletal/cardiac muscle; α6β1 binds multiple laminin isoforms (e.g., laminin-111, -511) | [99-101] |
| RGD-binding integrins | α5β1, αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, α8β1, αIIbβ3 | Fibronectin, fibrillin, fibrinogen, vitronectin, osteopontin (RGD motif) | Largest group; recognize the conserved RGD sequence; important in cell adhesion, migration, and angiogenesis | [67,102] |
| Leukocyte adhesion integrins | α4β1, α9β1, αLβ2, αMβ2, αXβ2, αDβ2, αEβ7, α4β7 | ICAMs, VCAM-1, MAdCAM-1, iC3b, fibrinogen, E-cadherin | Mediate immune cell adhesion and migration; β2 integrins also bind complement fragments (iC3b) and fibrinogen | [46,69] |
1. Arginine-glycine-aspartic acid (RGD)-binding integrins (the largest group), such as αvβ3, α5β1, and αIIbβ3, which recognize RGD motif in ECM components like fibronectin, fibrillin, and fibrinogen.
2. Leukocyte adhesion integrins, including β2 integrins (e.g., αLβ2, αMβ2) and members of the α4/α9/αE subfamily (e.g., α4β1, α4β7, α9β1, αEβ7), which mediate adhesion and migration of immune cells.
3. Collagen-binding integrins (e.g., α1β1, α2β1, α10β1, α11β1), which bind to collagen fibers through the GFOGER-like sequence and rely on the organizational structure of collagen fibers.
4. Laminin-binding integrins (e.g., α3β1, α6β1, α7β1, α6β4), which interact with laminin in the basement membrane.
Notably, some collagen-binding integrins (e.g., α1β1, α2β1, α10β1) also show affinity for laminin, suggesting potential functional overlap[31]. Additionally, integrin expression is tightly regulated by tissue specificity and developmental stage. Apart from canonical ECM mediators, integrins also interact with non-ECM ligands, including pathogen-derived surface proteins, growth factors, hormones, and bioactive compounds[105-107].
The function of integrins depends on maintaining a delicate balance between their active and inactive states through various mechanisms, including protein-protein interactions, conformational changes, and transport[108,109]. In their initial state, integrins exist on the cell surface in an inactive, low-affinity conformation. Integrin activation is typically triggered by intracellular signals, a process known as "inside-out" signaling. During this process, proteins like talin and kindlin bind to the intracellular tail of the β-integrin subunit, disrupting the transmembrane structure and inducing a conformational change in the integrin to form a high-affinity, extended state. In this state, integrins bind specific ECM ligands like fibronectin, collagen, or laminin[110-112].
After ligand binding, integrins aggregate and initiate “outside-in” signaling, a more complex and tightly regulated process. The intracellular domains of aggregated integrins serve as scaffolds for recruiting various adaptor proteins, including talin, vinculin, paxillin, and FAK[113-115]. These proteins typically contain phosphotyrosine-binding domain or four-point-one, ezrin, radixin, moesin domain domains that recognize phosphorylation sites or structural changes at adhesion sites[116-118]. The resulting large protein complexes, known as focal adhesion complexes (FACs), act as central hubs for signal transduction, activating downstream pathways such as FAK-Src, PI3K-Akt, and MAPK. These signaling pathways regulate diverse cellular functions such as adhesion, migration, proliferation, survival, and differentiation[119-121].
Notably, integrin activation in PH is not solely regulated by biochemical cues but is strongly influenced by the mechanical stiffness of the ECM. Progressive ECM remodeling in PH, characterized by excessive collagen and fibronectin deposition, leads to increased matrix stiffness, which enhances mechanical force transmission across integrin-ECM bonds. This elevated mechanical tension stabilizes integrins in their active, extended conformation, promotes integrin clustering, and amplifies outside-in signaling even in the absence of strong inside-out activation. Consequently, mechanosensitive pathways such as FAK, YAP/TAZ, and TGF-β signaling are persistently activated, driving smooth muscle cell proliferation, endothelial dysfunction, and fibrosis. While these cellular responses further exacerbate ECM remodeling and stiffening, forming a vicious cycle that sustains pathological vascular remodeling in PH.
The complex signaling network formed by integrins and their associated proteins illustrates the multifunctionality of integrins in mediating dynamic interactions between cells and the ECM, and in responding to biochemical and mechanical signals within the microenvironment[20]. Recent studies have further revealed that integrins not only play a key role in cell adhesion and migration, but also participate in many physiological and pathological processes by influencing cell morphology, metabolism, proliferation, apoptosis and other processes, especially playing an important role in the progression of diseases such as PH[111] [Figure 1].
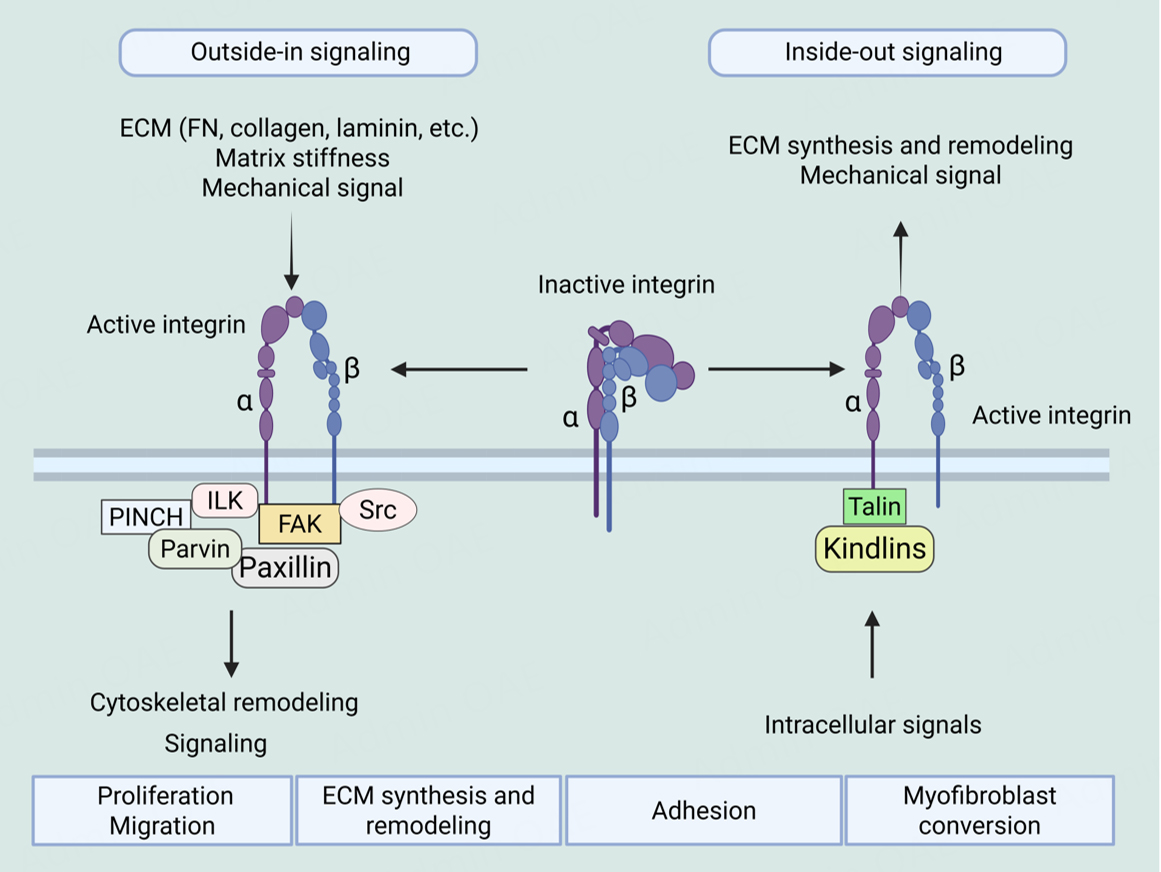
Figure 1. Integrin bidirectional signaling - integrin signaling from the outside in and inside out. In outside-in signaling, abnormal deposition of ECM components, alterations in matrix stiffness, and mechanical stimulation lead to integrin activation and recruitment of adaptor proteins to form integrin adhesion complexes, which trigger cytoskeletal remodeling and conduct or regulate downstream signaling cascades (from outside-in signaling). In inside-out signaling, intracellular signaling activation stimulates talin or kindlin binding to the cytoplasmic tail of integrins, inducing conformational changes in integrins and enhancing their affinity for ECM ligands. ECM: Extracellular matrix, ILK: integrin-linked kinase, FAK: focal adhesion kinase. Created in BioRender. He, S. (2026) https://BioRender.com/1o25dcs.
INTEGRINS AND PULMONARY HYPERTENSION: STRUCTURE, FUNCTION, AND SIGNALING MECHANISMS
The unique heterodimeric structure and conformational plasticity of integrin lay the foundation for their dual role as adhesion receptors and mechanosensors. Importantly, these structural features are not merely static properties but are tightly linked to intracellular signaling events. Understanding how integrin conformational shifts are translated into biochemical signals is therefore essential for elucidating their role in pulmonary vascular remodeling. In this section, we outline the major integrin-mediated signaling pathways that have been implicated in the pathogenesis of PH.
In PH, pulmonary vascular lesions are closely associated with abnormal deposition of ECM components[28]. Under stimuli such as hypoxia, inflammation, shear stress, and genetic predisposition, the pulmonary vascular microenvironment undergoes significant changes, manifesting as abnormal deposition of ECM components like fibronectin, collagen, and tenascin-C, leading to matrix stiffening and increased rigidity[122-124]. This pathological ECM remodeling provides favorable conditions for the overexpression and sustained activation of integrins.
In PH, pulmonary vascular lesions are closely associated with abnormal deposition of ECM components[28]. Under stimuli such as hypoxia, inflammation, shear stress, and genetic predisposition, the pulmonary vascular microenvironment undergoes significant changes, manifesting as abnormal deposition of ECM components like fibronectin, collagen, and tenascin-C, leading to matrix stiffening and increased rigidity[122-124]. This pathological ECM remodeling provides favorable conditions for the overexpression and sustained activation of integrins.
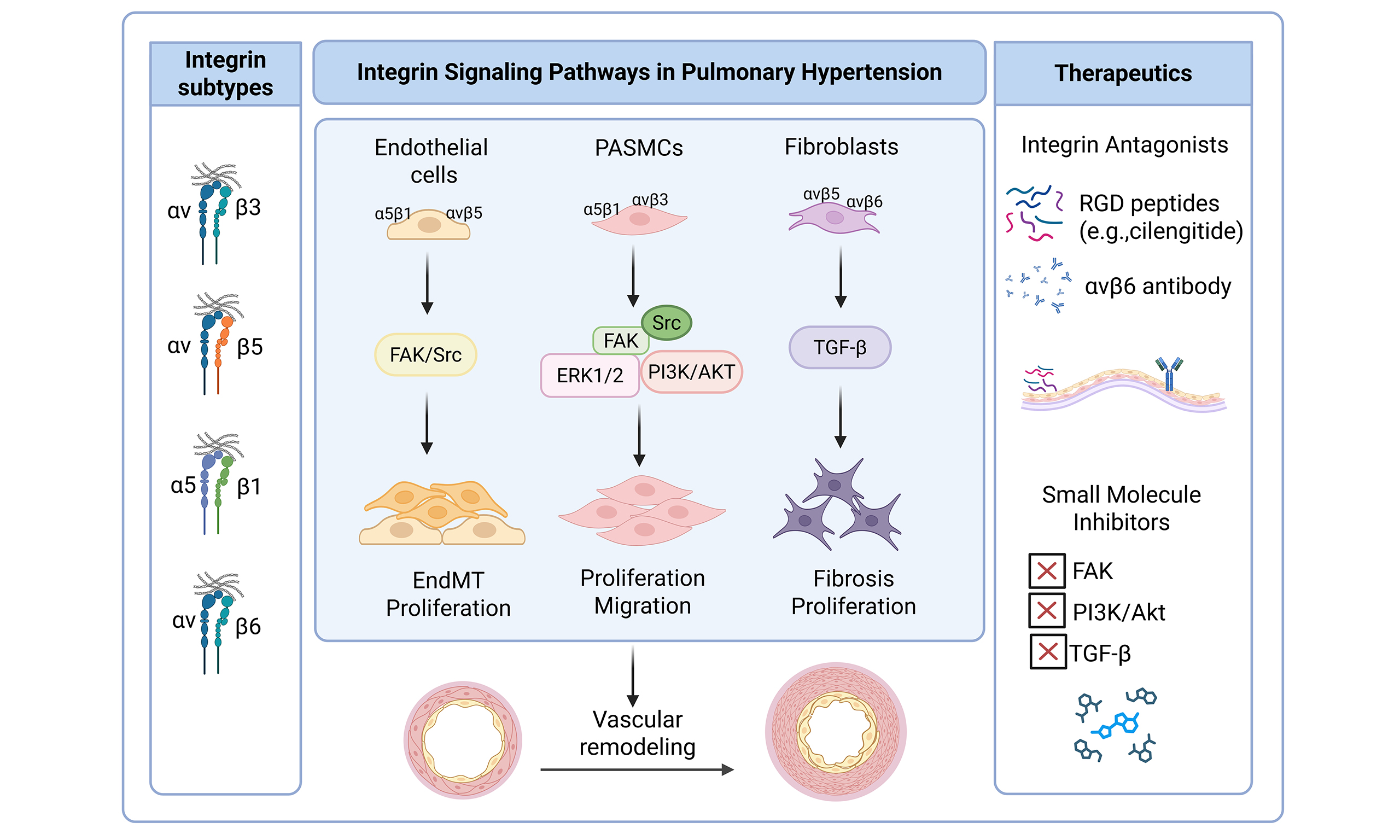
Studies have shown that in patients with PH and animal models, abnormally upregulated integrin subtypes such as α5β1, αvβ3, and αvβ5 are closely related to pulmonary vascular remodeling[22,125]. However, the expression and dysregulation of these integrins may vary across different PH subtypes. For example, in PAH, characterized by endothelial dysfunction and excessive smooth muscle cell proliferation, αvβ3 and α5β1 are particularly prominent, promoting cell proliferation and resistance to apoptosis[126]. In contrast, in CTEPH, where fibrotic occlusion of large pulmonary arteries predominates, integrin β2 (ITGB2) is increasingly implicated. Recent reports have found that ITGB2 is upregulated in platelets/immune cells in patients with CTEPH, which is related to the formation of neutrophil extracellular trap (NET) and the maintenance of inflammation, which may indirectly promote thrombus solidification and the progression of the disease course[127]. In addition, hypoxia—a key environmental driver of PH—further modulates integrin expression across subtypes via the HIF-1α signaling pathway[128], thereby directly linking external stress to integrin-mediated vascular remodeling. These subtype-specific patterns underscore the need for precision therapeutic strategies that target the most relevant integrin pathways according to the underlying etiology of PH.
Upon activation, integrin not only mediates the physical anchorage of cells to the ECM but more importantly, through the assembly of FACs, initiates a series of complex intracellular signaling cascades[31]. These signaling pathways play a crucial role in the pathological processes of PH, promoting abnormal proliferation, apoptosis resistance, migration, phenotype transformation, and inflammatory responses of PASMCs and ECs, ultimately leading to pulmonary vascular remodeling
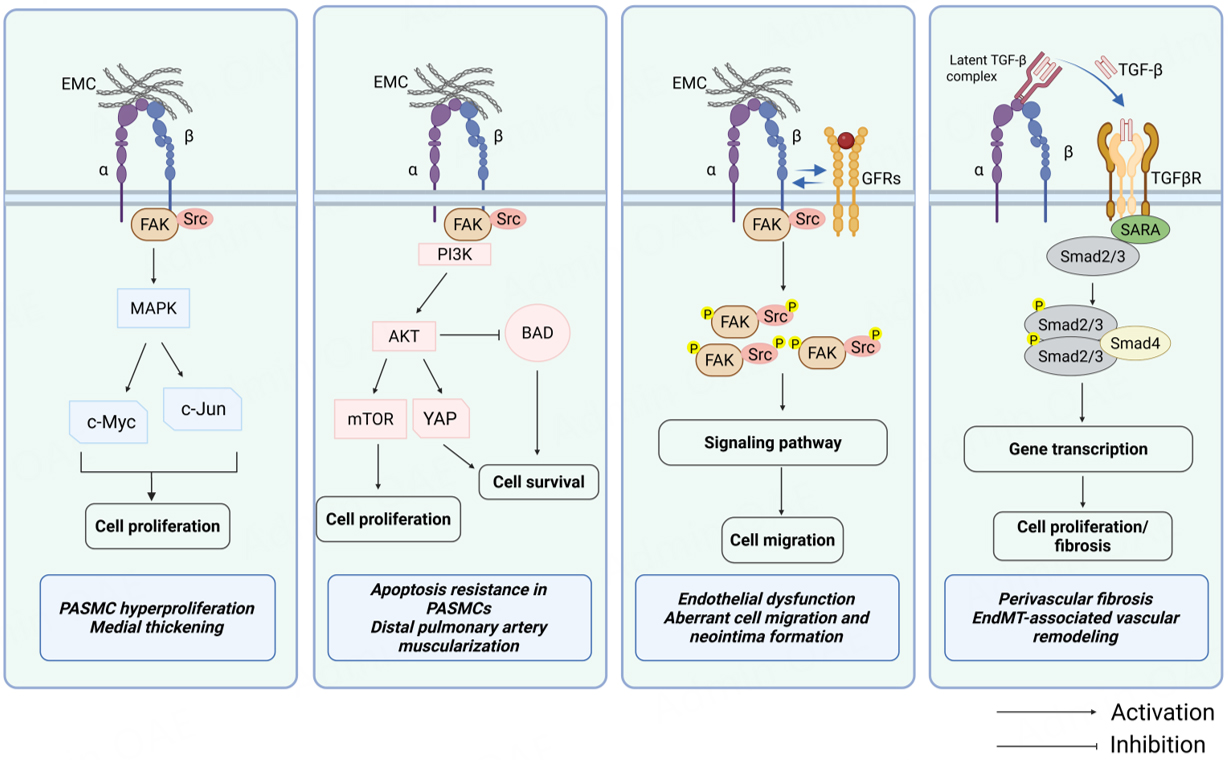
Figure 2. Integrin-mediated signaling pathways in PH. Integrin engagement activates multiple downstream signaling cascades that contribute to pulmonary vascular remodeling. (Left to right) MAPK pathway: FAK/Src-mediated activation of MAPK promotes transcription factors (c-Myc, c-Jun) driving PASMC and PAEC proliferation. PI3K/AKT pathway: integrin-FAK signaling activates PI3K/AKT, which enhances cell proliferation (via mTOR and YAP) and cell survival (via inhibition of BAD and FOXO-mediated pro-apoptotic signaling). Cross-talk with GFRs: Integrins cooperate with GFRs, amplifying downstream pathways and promoting migration and proliferation. TGF-β signaling: integrins mediate activation of latent TGF-β complexes, enabling TGF-β receptor signaling through Smad2/3-Smad4, which regulates cell proliferation and fibrosis. PH: Pulmonary hypertension, GFRs: growth factor receptors, FAK: focal adhesion kinase. Created in BioRender. He, S. (2026) https://BioRender.com/swfm3ic.
FAK and Src: core signaling hubs
FAK is a non-receptor tyrosine kinase that plays a central role in integrin signaling. When ECM proteins bind to integrins on the cell surface, integrins cluster and activate FAK. This activation triggers FAK to phosphorylate itself at tyrosine 397 (Tyr397). Phosphorylated Tyr397 then serves as a high-affinity binding site for Src family kinases—another group of tyrosine kinases[116]. Once bound, FAK and Src form a signaling complex that further phosphorylates several downstream proteins, such as paxillin, p130Cas, and talin. These proteins help regulate cytoskeletal rearrangement, adhesion dynamics, and cell migration[129]. Furthermore, the FAK-Src pathway activates pro-survival and proliferative pathways such as Ras-MAPK and PI3K-Akt pathways[130,131].
In PH, FAK exhibits persistent phosphorylation and high activity in both PASMCs and PAECs, particularly in neointimal and plexiform lesion areas[132,133]. Studies in animal models have shown that gene knockout or pharmacological inhibition of FAK significantly inhibits PASMC proliferation and migration, alleviates vascular wall thickening, and reduces right ventricular systolic pressure, thus improving pulmonary vascular remodeling[134]. This indicates that FAK is not only a central node of integrin signaling but also a highly promising therapeutic target for PH.
PI3K/AKT pathway: promoting cell survival and anti-apoptosis
Integrin-FAK signaling activates PI3K, either directly or indirectly. This activation of PI3K catalyzes the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). The newly formed PIP3 then recruits and activates AKT[135,136]. Once activated, AKT exerts strong pro-survival and anti-apoptotic effects by phosphorylating a variety of downstream effector molecules. For instance, AKT phosphorylates and inhibits the pro-apoptotic protein Bad, causing it to dissociate from the Bcl-2 complex[137,138]; it also inhibits the nuclear translocation of the transcription factor FOXO, thereby downregulating the expression of pro-apoptotic genes (such as Bim and FasL)[139]. In addition, AKT also promotes protein synthesis and cell growth by activating the mTOR pathway[140].
In PH patients and animal models, phosphorylation of AKT is significantly elevated, particularly in PASMCs. This persistent activation endows the cells with resistance to apoptotic stimuli (such as hypoxia and oxidative stress), contributing to excessive cell proliferation and medial thickening[141,142]. Studies suggest that inhibiting the PI3K/AKT pathway induces PASMC apoptosis and reverses vascular remodeling, suggesting a key role of this pathway in maintaining the homeostasis of PASMCs[143].
MAPK pathway: regulating proliferation and inflammatory response
Integrins also activate the MAPK family, including ERK1/2, JNK, and p38 MAPK, all of which regulate cell proliferation, stress response, and inflammation.
• ERK1/2 pathway: Integrins activate the FAK-Ras-Raf-MEK cascade to promote cell cycle progression (e.g., upregulating cyclin D1, inhibiting p27), driving abnormal proliferation of PASMCs and ECs. In hypoxic PH models, sustained activation of ERK is closely associated with vascular wall thickening[144,145].
• p38 MAPK pathway: This pathway is induced in response to inflammation and stress stimuli and is involved in the production and secretion of pro-inflammatory factors (e.g., IL-6, TNF-α), enhancing the local inflammatory response[146].
• JNK pathway: Activated under conditions of oxidative stress and mechanical tension, JNK regulates the balance between cell apoptosis and survival, and in late-stage PH, it may be involved in right ventricular remodeling[147].
The synergistic effect of these three pathways enables integrins to not only regulate pulmonary vascular structure but also participate in chronic inflammatory responses in PH.
Cross-talk with growth factor receptors: signal amplification and synergy
There is extensive "cross-talk" between integrins and growth factor receptors (GFRs), forming a synergistic signaling network that significantly enhances pathological signal output. This cross-talk occurs through two main mechanisms:
• Transactivation: Integrin aggregation promotes the phosphorylation of GFRs [e.g., platelet-derived GFRs (PDGFRs); epidermal growth factor receptors (EGFRs); vascular endothelial growth factor receptor (VEGFR)], initiating downstream signaling even in the absence of ligands. For instance, αvβ3 integrin mediates Src-dependent tyrosine phosphorylation of PDGFR, enhancing its sensitivity to platelet-derived growth factors (PDGF)[148].
• Co-localization and complex formation: Integrins and GFRs may form physical complexes on the cell membrane, efficiently, effectively recruiting and activating signaling molecules. For example, co-aggregation of αvβ3 with PDGFR significantly enhances the activation of MAPK and PI3K pathways, synergistically driving excessive proliferation and migration of PASMCs[149,150].
This integrin-GFR synergy explains why single-target therapies (e.g., PDGF inhibition alone) are often ineffective in clinical practice and suggests that combined inhibition of integrin and growth factor pathways could be more effective.
Regulation of TGF-β activation: linking mechanical signals to fibrosis
TGF-β is a key pleiotropic cytokine that drives fibrosis, endothelial-mesenchymal transition (EndMT), and ECM deposition in PH[151-153]. TGF-β usually exists in an inactive "latent complex" form in the ECM and remains dormant by binding to latent TGF-β-binding proteins (LTBPs)[154]. Mechanical activation of latent TGF-β represents a key regulatory node linking matrix remodeling to profibrotic signaling.
Integrins αvβ6 and αvβ8 recognize these latent complexes through their RGD motif and, under mechanical stress, induce conformational changes that release biologically active TGF-β[155,156]. While early work established this activation mechanism, recent preclinical studies have substantially advanced our understanding of its pathological relevance. Emerging evidence published after 2023 demonstrates that genetic or pharmacological blockade of αvβ6 and αvβ8 integrins effectively suppresses TGF-β activation in vivo, leading to marked attenuation of fibrotic remodeling and mesenchymal transition programs[25,26,157].
In the context of PH, αvβ6 and αvβ8 expression is markedly upregulated in perivascular fibroblasts and dysfunctional pulmonary endothelial cells, creating a permissive microenvironment for sustained TGF-β activation[155]. Recent experimental studies further suggest that integrin-dependent TGF-β activation not only promotes fibroblast-to-myofibroblast transformation and the secretion of collagen and fibronectin but also induces EndMT in endothelial cells, granting them the ability to migrate and synthesize ECM, thereby directly contributing to perivascular fibrosis and vascular wall thickening[157,158]. Although these mechanistic insights are primarily derived from previous preclinical models, they provide strong support for a conserved integrin-TGF-β axis that links mechanical cues to fibrotic remodeling in pulmonary vascular disease.
These findings indicate that integrin-mediated TGF-β activation plays a critical role in PH, acting as a key bridge connecting mechanical microenvironment changes to tissue fibrosis. Through this mechanism, integrins not only promote vascular fibrosis but also accelerate PH progression, providing a potential new direction for therapeutic strategies targeting integrin and TGF-β pathways.
Collectively, accumulating evidence indicates that distinct integrin subtypes are differentially expressed across pulmonary vascular cell populations and contribute to multiple facets of pulmonary vascular remodeling, including smooth muscle cell hyperplasia, endothelial dysfunction, inflammation, and fibrosis. To provide an integrated overview, the major integrin subtypes implicated in PH, along with their predominant cellular expression, pathological roles, therapeutic relevance, and correspondence with distinct PH phenotypes, are summarized in Table 3.
Key integrin subtypes in pulmonary hypertension: cellular distribution, pathological roles, and therapeutic relevance
| Integrin subtype | Primary cell types in PH | Key pathological roles | Representative signaling pathways | Therapeutic relevance | Main PH subtype association |
| α5β1 | PASMCs, PAECs | Drives PASMC proliferation and migration; promotes medial hypertrophy and ECM stiffening | FAK-Src, PI3K/Akt, MAPK; fibronectin-dependent mechanotransduction | Emerging target; α5β1 blockade attenuates vascular remodeling in preclinical PH models | PAH[22] |
| αvβ3 / αvβ5 | PASMCs, PAECs (plexiform lesions) | Promotes pathological angiogenesis, apoptosis resistance, and hyperproliferation | FAK-ERK, PI3K/Akt; cross-talk with PDGFR/VEGFR | RGD-based inhibitors (e.g., cilengitide); translational lessons from oncology | PAH[126,159], Congenital Heart Disease-Associated PAH[125] |
| αvβ6 | Perivascular fibroblasts, dysfunctional PAECs | Activates latent TGF-β; promotes EndMT-like phenotypic switching and perivascular fibrosis | Mechanical TGF-β activation; Smad2/3 signaling | Small-molecule and peptide inhibitors under investigation in fibrotic diseases | PAH[160]; pulmonary inflammation and fibrosis[155] |
| αvβ8 | Endothelial cells, fibroblasts | Regulates local TGF-β bioavailability; contributes to vascular fibrosis and inflammatory remodeling | TGF-β/Smad signaling; ECM-integrin feedback | Dual αvβ6/αvβ8 blockade proposed to suppress pathological TGF-β activation | PAH[160] |
| α1β1 / α2β1 | PASMCs, fibroblasts | Mediates collagen sensing; reinforces ECM-driven vascular stiffening | Collagen-dependent integrin signaling | Largely unexplored in PH; potential modulators of matrix-cell feedback | Unknown |
| β1 integrin (general) | PASMCs, PAECs, fibroblasts | Central mechanosensor sustaining proliferative and fibrotic signaling loops | FAK-YAP/TAZ-Hippo axis | Broad mechanotransduction target with translational potential | PAH[161], Hypoxic pulmonary hypertension (PH)[162] |
THERAPEUTIC TARGETING OF INTEGRINS
Integrin inhibitors as potential therapeutic strategies for PAH
Given the central role of integrins in extracellular matrix remodeling, mechanotransduction, and pathological vascular signaling in PH, targeting specific integrin subtypes or their downstream effectors has emerged as a promising therapeutic approach. By directly modulating integrin activity, we can disrupt the aberrant activation of key downstream signaling pathways—namely FAK, TGF-β, and YAP/TAZ—which converge on fundamental processes driving pulmonary vascular remodeling, including abnormal cell proliferation, resistance to apoptosis, dysregulated mechanotransduction, and fibrosis. This approach offers a mechanistically grounded strategy not merely to alleviate symptomatic vasoconstriction, but to reverse the underlying structural pathology of PH. Preclinical studies have evaluated several integrin-directed agents—such as RGD-mimetic peptides and small-molecule inhibitors—with encouraging results in animal models of PH. This section reviews current advances and challenges in the development of integrin-targeted therapies for PH, and the major integrin-targeting therapeutic candidates discussed are summarized in
Summary of integrin-targeting therapeutic candidates for pulmonary hypertension (PH)
| Drug name | Target integrin | Current stage of development | Key findings & potential relevance to PAH | References |
| MRT1 | α5β1 | Preclinical | Reverses established pulmonary vascular remodeling and improves right heart function in MCT and SuHx PH models; efficacy comparable or superior to FDA-approved PAH drug Sotatercept | [22,163] |
| Volociximab | α5β1 | Phase I (oncology) | Monoclonal antibody with acceptable tolerability in cancer clinical trials; identified as a potential disease-modifying target for PAH, pending specialized PAH clinical validation | [164] |
| ATN-161 | α5β1 | Phase I (oncology) | Non-RGD pentapeptide integrin antagonist; confirmed safety in oncology Phase I trials; alleviates vascular remodeling in cardiovascular disease models, with translational potential for PAH | [165,166] |
| Cilengitide | αvβ3/αvβ5 | Phase I/II (oncology) | Cyclic RGD peptide; manageable safety profile in multiple oncology Phase I/II trials; promising candidate for PAH, especially for patients with fibrotic phenotypes | [167-170] |
| MK-0429 | Pan-αv integrins | Preclinical | Exerts antifibrotic effects in renal and pulmonary fibrosis models; untested in PAH but holds translational value for PAH with fibrotic features | [171,172] |
| GSK3008348 | αvβ6 | Clinical (IPF; inhaled) | Effectively binds target and inhibits TGF-β pathway in IPF patients; potential relevance for PAH patients with concomitant fibrosis | [173] |
| BG00011 (STX-100) | αvβ6 | Phase I (IPF) | Monoclonal antibody with anti-fibrotic effects in IPF models; good safety and tolerability in Phase I trials; may benefit PAH patients with concurrent fibrosis | [174,175] |
| Bexotegrast (PLN-74809) | Dual αvβ6/αvβ1 | Phase I (IPF) | Dual integrin inhibitor with anti-fibrotic activity in IPF; favorable safety profile in Phase I trials; potential therapeutic value for PAH with concomitant fibrosis | [176] |
| EDIL3-αvβ3 Axis Inhibitors | EDIL3-αvβ3 ligand-receptor axis | Preclinical | Blockade downregulates ERK1/2 activation, inhibits PASMC proliferation and migration, and alleviates pulmonary vascular remodeling; provides proof-of-concept for ligand-receptor | [159] |
α5β1 integrin inhibitors
Among various subtypes, α5β1 has been the most extensively studied target. Preclinical studies have shown that blocking α5β1 function with research-grade antibodies or small molecule inhibitors like Myocardin-related transcription factor 1 in both monocrotaline (MCT) and Sugen/hypoxia (SuHx) models reverses existing pulmonary vascular remodeling and improves right heart function[22]. Notably, these effects are comparable or even superior to Sotatercept, an FDA-approved drug for adult PAH[163]. Moreover, the α5β1 monoclonal antibody, volociximab, has undergone early clinical trials in cancer patients[164], showing acceptable tolerability, suggesting that α5β1 is a potential target for disease-modifying therapy. However, specialized clinical validation in PAH patients is still needed.
Small molecule and peptide integrin inhibitors
Various small molecule and peptide integrin inhibitors have shown promise for repositioning. ATN-161, a non-RGD pentapeptide that antagonizes α5β1, has demonstrated safety in phase I oncology trials and alleviates vascular remodeling in cardiovascular disease models[165,166]. Cilengitide, a cyclic RGD peptide targeting αvβ3/αvβ5, has completed several phase I/II clinical trials in oncology with manageable safety[167-170]. MK-0429, a pan-inhibitor of αv integrins, has shown antifibrotic effects in renal and pulmonary fibrosis models[171,172]. Although these molecules have not been tested in PAH patients, their pharmacological characteristics suggest significant translational potential, particularly in PAH patients with fibrotic phenotypes.
αvβ6/αvβ1 inhibitors
Some integrin inhibitors developed for fibrotic diseases may be relevant to PAH with concomitant fibrosis. GSK3008348, an inhaled αvβ6 inhibitor, has been shown to effectively bind the target and inhibit the TGF-β pathway in idiopathic pulmonary fibrosis (IPF) patients[173]. BG00011 (STX-100, αvβ6 monoclonal antibody) and Bexotegrast (PLN-74809, dual αvβ6/αvβ1 inhibitor) have shown anti-fibrotic effects in IPF, with good safety and tolerability in phase I trials[174-176]. Though these drugs have not been tested in PAH, they may have potential value in PAH patients with concurrent fibrosis.
Ligand-receptor axis intervention
Recent studies have identified the EDIL3-αvβ3 axis as a critical driver of PASMC proliferation and migration. Blocking this interaction downregulates ERK1/2 activation and alleviates vascular remodeling[159]. This provides proof of concept for cutting off pathological signals at the ligand-receptor interface.
Taken together, despite compelling preclinical evidence supporting integrin-targeted interventions in pulmonary vascular remodeling, there is currently a lack of Phase II/III clinical trial data evaluating integrin inhibitors in PAH patients. Future translational efforts should prioritize carefully designed early-phase clinical trials incorporating biomarker-based patient stratification, disease-stage-specific enrollment, and mechanistic endpoints to bridge this critical gap between bench and bedside.
CHALLENGES AND PROSPECTS
With the deepening of research into the pathogenesis of PH, the role of the ECM and mechanotransduction systems in pulmonary vascular remodeling has received increasing attention[15]. As a key molecule linking cells to the ECM, integrins play an important role in the pathology of PH[22]. Although integrin signaling has been shown to play a crucial role in PH pathogenesis, the current challenge lies in how to translate these findings into effective clinical treatments.
There are many members of the integrin family, and their functions and expression differ significantly across different tissues[31]. Therefore, accurately identifying specific integrin subtypes associated with PH and developing highly selective inhibitors is critical for avoiding off-target effects and reducing side effects. Additionally, it is necessary to gain a deeper understanding of the specific roles of each subtype in different types of PH to achieve precision therapy.
While several integrin inhibitors have made progress in oncology and fibrotic diseases, clinical validation in PH is still in the early stages[177,178]. One major barrier to clinical translation in PH is efficacy concerns, as integrin inhibitors have shown limited success in targeting vascular remodeling in PH. Furthermore, biomarker limitations in PH complicate patient stratification, making it difficult to identify those most likely to benefit from integrin-targeted therapies. Off-target effects, particularly in the cardiovascular system, may also contribute to safety concerns. These challenges highlight the need for further research into integrin inhibitor efficacy in PH and the development of reliable biomarkers to guide patient selection for future trials.
Importantly, the limited success of certain integrin inhibitors in oncology trials offers valuable insights for their potential application in PH. For instance, Cilengitide—a cyclic RGD peptide targeting αvβ3 and αvβ5—exhibited promising anti-angiogenic activity in preclinical tumor models yet failed to deliver significant survival benefits in Phase III clinical trials for glioblastoma. Several key factors have been implicated in this outcome, including inadequate biomarker-guided patient stratification, functional redundancy among integrin subtypes, and paradoxical pro-angiogenic effects at suboptimal dosing concentrations[179]. These clinical experiences hold profound relevance for PH translational research. In contrast to oncology, PH is a chronic progressive disorder requiring long-term therapeutic intervention, where safety profiles, rational dosing strategies, and sustained target engagement are of particular importance. Furthermore, the inherent heterogeneity of PH suggests that only distinct patient subgroups—such as those with marked integrin overexpression or ECM-driven vascular remodeling—are likely to derive clinical benefit from integrin-targeted therapies[180]. Therefore, successful translation of such agents in PH will likely hinge on improved biomarker-based patient selection, disease-stage-specific therapeutic intervention, and meticulous optimization of dosing regimens to mitigate off-target effects or compensatory signaling pathways.
Systemic administration of integrin inhibitors may lead to severe side effects, particularly in the cardiovascular system[181]. Therefore, improving the delivery efficiency of drugs to the lungs and reducing systemic side effects is key to achieving clinical translation. Nanotechnology, liposomes, and other targeted delivery systems provide new approaches to solving this problem. For example, nanoparticles surface-modified with RGD peptides or anti-PECAM antibodies actively targets activated endothelial cells, enabling localized drug accumulation[182,183]. Moreover, inhaled formulations (such as nebulized inhalers) also represent a potential delivery method, directly acting on the pulmonary lesions and reducing systemic exposure[173].
Biomarkers and treatment responses in different types of PH patients show significant differences. For instance, some patients may exhibit overexpression of specific integrin subtypes, while others may rely on different signaling pathways (e.g., PDGF, VEGF)[23,126]. Developing individualized treatment plans based on molecular characteristics remains an urgent challenge.
Looking ahead, the development of imaging-based and circulating biomarkers reflecting integrin expression or activation status may provide powerful tools for patient stratification in PH. While direct clinical evidence supporting circulating soluble integrin ectodomains as validated biomarkers remains limited, increasing attention has focused on ECM-derived peptides—such as collagen degradation neo-epitopes and matrikines—which reflect pathological matrix turnover and vascular fibrosis. Several studies in PH and related fibrotic diseases have demonstrated that these ECM fragments correlate with disease severity and hemodynamic impairment[184-186]. In parallel, integrin-targeted molecular imaging approaches, including αvβ3-directed PET tracers, are being explored to enable non-invasive assessment of regional vascular remodeling and integrin activation in vivo, highlighting an emerging translational framework for biomarker-guided precision therapy.
Despite the multiple challenges currently faced in the study of integrins in PH, their emerging role as a therapeutic target offers unprecedented treatment potential. Through further basic research and clinical trials, we expect to see integrin-targeted therapies become a new option for PH treatment. By combining modern molecular biology techniques and drug delivery systems, integrin-targeted therapies not only have the potential to slow vascular remodeling but also reverse the structural damage caused by PH, offering patients better prognosis.
CONCLUSION
Integrin signaling pathways play a central role in the pathogenesis of PH, driving vascular remodeling through their impact on cell proliferation, survival, inflammation, and ECM dynamics. Dysregulation of integrin expression and abnormal activation of downstream signaling pathways (such as FAK, PI3K/Akt, and MAPK) create a favorable environment for disease progression. While current treatments mainly target vasoconstriction, integrin-targeted therapies offer a strategy to address the structural basis of PH. Integrin-targeted strategies not only alleviate vasoconstriction but may also reverse structural remodeling, representing a paradigm shift in PH therapeutics. Although clinical translation faces obstacles, advances in selective inhibitors and delivery systems hold promising potential. A deeper understanding of integrin biology in PH is essential for developing effective disease-modifying therapies.
DECLARATIONS
Acknowledgments
The graphical abstract was created using BioRender.com (Created in BioRender. He, S. (2026) https://BioRender.com/az449wg).
Authors’ contributions
Conceptualized the manuscript: He S, Bian JS
Wrote the manuscript text and made a visualization: He S
Reviewed the manuscript: Nie X, Bian JS
Completed the formal analysis: Zhang J, Li Y, Liu H
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version 4.0, released 2023-11-15) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Grant No. 82570080 to Nie X) and Shenzhen Science and Technology Program, Shenzhen, China (Grant No. GJHZ20240218111401002 to Bian JS).
Conflicts of interest
Bian JS is an Editorial Board Member of the journal Vessel Plus, but was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
2. Boucly A, Gerges C, Savale L, et al. Pulmonary arterial hypertension. Presse Med. 2023;52:104168.
3. Humbert M, Guignabert C, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53:1801887.
4. Kovacs G, Bartolome S, Denton CP, et al. Definition, classification and diagnosis of pulmonary hypertension. Eur Respir J. 2024;64:2401324.
6. Sun L, Tian H, Liu J, et al. The role of the pulmonary vascular microenvironment in chronic thromboembolic pulmonary hypertension. Pulm Circ. 2025;15:e70118.
7. Ruaro B, Baratella E, Caforio G, et al. Chronic thromboembolic pulmonary hypertension: an update. Diagnostics. 2022;12:235.
8. Mocumbi A, Humbert M, Saxena A, et al. Publisher Correction: pulmonary hypertension. Nat Rev Dis Primers. 2024;10:5.
9. Naeije R, Richter MJ, Rubin LJ. The physiological basis of pulmonary arterial hypertension. Eur Respir J. 2022;59:2102334.
10. Dai J, Chen H, Fang J, Wu S, Jia Z. Vascular remodeling: the multicellular mechanisms of pulmonary hypertension. Int J Mol Sci. 2025;26:4265.
11. Gallardo-vara E, Ntokou A, Dave JM, Jovin DG, Saddouk FZ, Greif DM. Vascular pathobiology of pulmonary hypertension. J Heart Lung Transplant. 2023;42:544-52.
12. Guignabert C, Aman J, Bonnet S, et al. Pathology and pathobiology of pulmonary hypertension: current insights and future directions. Eur Respir J. 2024;64:2401095.
13. Christou H, Khalil RA. Mechanisms of pulmonary vascular dysfunction in pulmonary hypertension and implications for novel therapies. Am J Physiol Heart Circ Physiol. 2022;322:H702-24.
14. Shen Y, Goncharov DA, Pena A, et al. Cross-talk between TSC2 and the extracellular matrix controls pulmonary vascular proliferation and pulmonary hypertension. Sci Signal. 2022;15:eabn2743.
15. Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2018;315:H1322-31.
16. Liu F, Haeger CM, Dieffenbach PB, et al. Distal vessel stiffening is an early and pivotal mechanobiological regulator of vascular remodeling and pulmonary hypertension. JCI Insight. 2016;1:e86987.
17. Mamazhakypov A, Maripov A, Sarybaev AS, Schermuly RT, Sydykov A. Osteopontin in pulmonary hypertension. Biomedicines. 2023;11:1385.
18. Chastney MR, Kaivola J, Leppänen V, Ivaska J. The role and regulation of integrins in cell migration and invasion. Nat Rev Mol Cell Biol. 2024;26:147-67.
22. Lemay S, Montesinos MS, Grobs Y, et al. Exploring integrin α5β1 as a potential therapeutic target for pulmonary arterial hypertension: insights from comprehensive multicenter preclinical studies. Circulation. 2025;151:1162-83.
23. Umesh A, Paudel O, Cao Y, Myers AC, Sham JS. Alteration of pulmonary artery integrin levels in chronic hypoxia and monocrotaline-induced pulmonary hypertension. J Vasc Res. 2011;48:525-37.
24. Suezawa T, Kanagaki S, Moriguchi K, et al. Disease modeling of pulmonary fibrosis using human pluripotent stem cell-derived alveolar organoids. Stem Cell Rep. 2021;16:2973-87.
25. Bellani S, Molyneaux PL, Maher TM, Spagnolo P. Potential of αvβ6 and αvβ1 integrin inhibition for treatment of idiopathic pulmonary fibrosis. Expert Opin Ther Targets. 2024;28:575-85.
26. Gasparri AM, Pocaterra A, Colombo B, et al. Blockade of αvβ6 and αvβ8 integrins with a chromogranin A-derived peptide inhibits TGFβ activation in tumors and suppresses tumor growth. J Exp Clin Cancer Res. 2025;44:88.
27. Bi Z, Zang G, Wang X, Tian L, Zhang W. Integrins and pulmonary fibrosis: pathogenic roles and therapeutic opportunities. Biomol Biomed. 2025;26:200-14.
28. Jandl K, Radic N, Zeder K, Kovacs G, Kwapiszewska G. Pulmonary vascular fibrosis in pulmonary hypertension - The role of the extracellular matrix as a therapeutic target. Pharmacol Ther. 2023;247:108438.
29. Yang Q, Chen P, He X, et al. Melk facilitates pulmonary artery smooth muscle cell proliferation and migration in pulmonary hypertension via modulation of YAP/TAZ signaling. Front Cell Dev Biol. 2025;13:1693346.
30. Zhao T, Huang Y, Zhu J, et al. Extracellular matrix signaling cues: biological functions, diseases, and therapeutic targets. MedComm. 2025;6:e70281.
31. Pang X, He X, Qiu Z, et al. Targeting integrin pathways: mechanisms and advances in therapy. Sig Transduct Target Ther. 2023;8:1.
32. Tamkun JW, Desimone DW, Fonda D, et al. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell. 1986;46:271-82.
33. Hynes RO. The emergence of integrins: a personal and historical perspective. Matrix Biol. 2004;23:333-40.
34. Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011;3:a004994.
35. Hemler ME, Jacobson JG, Brenner MB, Mann D, Strominger JL. VLA‐1: a T cell surface antigen which defines a novel late stage of human T cell activation. Eur J Immunol. 2005;15:502-8.
37. Hamaia SW, Pugh N, Raynal N, et al. Mapping of potent and specific binding motifs, GLOGEN and GVOGEA, for integrin α1β1 using collagen toolkits II and III. J Biol Chem. 2012;287:26019-28.
38. Zeltz C, Gullberg D. The integrin-collagen connection - a glue for tissue repair? J Cell Sci. 2016;129:653-64.
40. Ramovs V, Te Molder L, Sonnenberg A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2017;57-8:213-43.
42. Couvelard A, Bringuier A, Dauge M, et al. Expression of integrins during liver organogenesis in humans. Hepatology. 1998;27:839-47.
43. Lora JM, Rowader KE, Soares L, Giancotti F, Zaret KS. α3β1-integrin as a critical mediator of the hepatic differentiation response to the extracellular matrix. Hepatology. 1998;28:1095-104.
44. Kreidberg JA, Donovan MJ, Goldstein SL, et al. Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537-47.
45. Kim YY, Lim CS, Song YH, Ahnn J, Park D, Song WK. Cellular localization of α3β1 integrin isoforms in association with myofibrillogenesis during cardiac myocyte development in culture. Cell Adhes Commun. 2009;7:85-97.
46. Guenther C. β2-integrins - regulatory and executive bridges in the signaling network controlling leukocyte trafficking and migration. Front Immunol. 2022;13:809590.
49. Hou J, Yan D, Liu Y, Huang P, Cui H. The roles of integrin α5β1 in human cancer. Onco Targets Ther. 2020;13:13329-44.
50. Renner G, Noulet F, Mercier M, et al. Expression/activation of α5β1 integrin is linked to the β-catenin signaling pathway to drive migration in glioma cells. Oncotarget. 2016;7:62194-207.
51. Lv X, Li Z, Guan J, et al. Porcine hemagglutinating encephalomyelitis virus activation of the integrin α5β1-FAK-cofilin pathway causes cytoskeletal rearrangement to promote its invasion of N2a cells. J Virol. 2019;93:e01736-18.
52. Oh S, Kim J, Kim Y, et al. The extracellular matrix protein Edil3 stimulates osteoblast differentiation through the integrin α5β1/ERK/Runx2 pathway. PLoS ONE. 2017;12:e0188749.
53. López-luppo M, Catita J, Ramos D, et al. Cellular senescence is associated with human retinal microaneurysm formation during aging. Invest Ophthalmol Vis Sci. 2017;58:2832.
54. Di Maggio N, Martella E, Frismantiene A, et al. Extracellular matrix and α5β1 integrin signaling control the maintenance of bone formation capacity by human adipose-derived stromal cells. Sci Rep. 2017;7:44398.
55. Delwel GO, De Melker AA, Hogervorst F, et al. Distinct and overlapping ligand specificities of the alpha 3A beta 1 and alpha 6A beta 1 integrins: recognition of laminin isoforms. Mol Biol Cell. 1994;5:203-15.
56. Hayashi YK, Chou F, Engvall E, et al. Mutations in the integrin α7 gene cause congenital myopathy. Nat Genet. 1998;19:94-7.
57. Mayer U, Saher G, Fässler R, et al. Absence of integrin α7 causes a novel form of muscular dystrophy. Nat Genet. 1997;17:318-23.
58. Flintoff‐dye NL, Welser J, Rooney J, et al. Role for the α7β1 integrin in vascular development and integrity. Dev Dyn. 2005;234:11-21.
60. Zargham R. Tensegrin in context: dual role of α8 integrin in the migration of different cell types. Cell Adh Migr. 2014;4:485-90.
61. Nishimichi N, Tsujino K, Kanno K, et al. Induced hepatic stellate cell integrin, α8β1, enhances cellular contractility and TGFβ activity in liver fibrosis. J Pathol. 2021;253:366-73.
62. Guo Q, Furuta K, Lucien F, et al. Integrin β1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol. 2019;71:1193-205.
63. Honda M, Kimura C, Uede T, Kon S. Neutralizing antibody against osteopontin attenuates non-alcoholic steatohepatitis in mice. J Cell Commun Signal. 2020;14:223-32.
64. Lundgren-Åkerlund E, Aszòdi A. Integrin α10β1: a collagen receptor critical in skeletal development. Adv Exp Med Biol. 2014;19:61-71.
65. Camper L, Holmvall K, Wängnerud C, Aszódi A, Lundgren-Åkerlund E. Distribution of the collagen-binding integrin α10β1 during mouse development. Cell Tissue Res. 2001;306:107-16.
66. Popova SN, Barczyk M, Tiger C, et al. α 11β1 integrin-dependent regulation of periodontal ligament function in the erupting mouse incisor. Mol Cell Biol. 2023;27:4306-16.
67. Han Z, Ma Y, Cao G, et al. Integrin αVβ1 regulates procollagen I production through a non-canonical transforming growth factor β signaling pathway in human hepatic stellate cells. Biochem J. 2021;478:1689-703.
68. Reed NI, Jo H, Chen C, et al. The αvβ1 integrin plays a critical in vivo role in tissue fibrosis. Sci Transl Med. 2015;7:288ra79.
69. Mitroulis I, Alexaki VI, Kourtzelis I, Ziogas A, Hajishengallis G, Chavakis T. Leukocyte integrins: role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol Ther. 2015;147:123-35.
71. Schnitzler N, Haase G, Podbielski A, Lütticken R, Schweizer KG. A co-stimulatory signal through ICAM-β2 integrin-binding potentiates neutrophil phagocytosis. Nat Med. 1999;5:231-5.
72. Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393-400.
73. Jawhara S, Pluskota E, Cao W, Plow EF, Soloviev DA. Distinct effects of integrins αXβ2 and αMβ2 on leukocyte subpopulations during inflammation and antimicrobial responses. Infect Immun. 2017;85:e00644-16.
74. Guenther C, Faisal I, Fusciello M, et al. β2-integrin adhesion regulates dendritic cell epigenetic and transcriptional landscapes to restrict dendritic cell maturation and tumor rejection. Cancer Immunol Res. 2021;9:1354-69.
75. Miyazaki Y, Vieira-De-Abreu A, Harris ES, et al. Integrin αDβ2 (CD11d/CD18) is expressed by human circulating and tissue myeloid leukocytes and mediates inflammatory signaling. PLoS ONE. 2014;9:e112770.
76. Yakubenko VP, Yadav SP, Ugarova TP. Integrin αDβ2, an adhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood. 2006;107:1643-50.
77. Van Den Kerkhof DL, Van Der Meijden PE, Hackeng TM, Dijkgraaf I. Exogenous integrin αIIbβ3 inhibitors revisited: past, present and future applications. Int J Mol Sci. 2021;22:3366.
78. Kokubo T, Uchida H, Choi ET. Integrin αvβ3 as a target in the prevention of neointimal hyperplasia. J Vasc Surg. 2007;45:A33-8.
79. Bishop GG, Mcpherson JA, Sanders JM, et al. Selective αvβ3-receptor blockade reduces macrophage infiltration and restenosis after balloon angioplasty in the atherosclerotic rabbit. Circulation. 2001;103:1906-11.
80. Guermonprez P, Valladeau J, Zitvogel L, Théry C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621-67.
81. Der Neut R, Krimpenfort P, Calafat J, Niessen CM, Sonnenberg A. Epithelial detachment due to absence of hemidesmosomes in integrin β4 null mice. Nat Genet. 1996;13:366-9.
82. Welser-alves JV, Boroujerdi A, Tigges U, Wrabetz L, Feltri ML, Milner R. Endothelial β4 integrin is predominantly expressed in arterioles, where it promotes vascular remodeling in the hypoxic brain. Arterioscle, Thromb Vasc Biol. 2013;33:943-53.
83. Porte J, Jenkins G, Tatler AL. Myofibroblast TGF-β activation measurement in vitro. Methods Mol Biol. 2021;2299:99-108.
84. Tatler AL, John AE, Jolly L, et al. Integrin αvβ5-mediated TGF-β activation by airway smooth muscle cells in asthma. J Immunol. 2011;187:6094-107.
85. Asano Y, Ihn H, Jinnin M, Mimura Y, Tamaki K. Involvement of αvβ5 integrin in the establishment of autocrine TGF-β signaling in dermal fibroblasts derived from localized scleroderma. J Investig Dermatol. 2006;126:1761-9.
86. Kumawat AK, Yu C, Mann EA, Schridde A, Finnemann SC, Mowat AM. Expression and characterization of αvβ5 integrin on intestinal macrophages. Eur J Immunol. 2018;48:1181-7.
87. Schiesser JV, Loudovaris T, Thomas HE, Elefanty AG, Stanley EG. Integrin αvβ5 heterodimer is a specific marker of human pancreatic beta cells. Sci Rep. 2021;11:8315.
88. Koivisto L, Bi J, Häkkinen L, Larjava H. Integrin αvβ6: structure, function and role in health and disease. Int J Biochem Cell Biol. 2018;99:186-96.
89. Madala SK, Korfhagen TR, Schmidt S, et al. Inhibition of the αvβ6 integrin leads to limited alteration of TGF-α-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2014;306:L726-35.
90. Ansar M, Jan A, Santos-cortez RLP, et al. Expansion of the spectrum of ITGB6-related disorders to adolescent alopecia, dentogingival abnormalities and intellectual disability. Eur J Hum Genet. 2015;24:1223-7.
91. White JB, Hu LY, Boucher DL, Sutcliffe JL. ImmunoPET imaging of αvβ6 expression using an engineered anti-αvβ6 Cys-diabody site-specifically radiolabeled with Cu-64: considerations for optimal imaging with antibody fragments. Mol Imaging Biol. 2017;20:103-13.
92. Li H, Huang S, Shi F, Gu Z, Zhang S, Wei J. α4β7 integrin inhibitors: a patent review. Expert Opin Ther Pat. 2018;28:903-17.
93. Arthos J, Cicala C, Nawaz F, et al. The role of integrin α4β7 in HIV pathogenesis and treatment. Curr HIV/AIDS Rep. 2018;15:127-35.
94. Fukui T, Fukaya T, Uto T, et al. Pivotal role of CD103 in the development of psoriasiform dermatitis. Sci Rep. 2020;10:8371.
95. Cepek KL, Shaw SK, Parker CM, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature. 1994;372:190-3.
96. Zundler S, Becker E, Spocinska M, et al. Hobit- and Blimp-1-driven CD4+ tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol. 2019;20:288-300.
97. Zhou M, Niu J, Wang J, et al. Integrin αvβ8 serves as a novel marker of poor prognosis in colon carcinoma and regulates cell invasiveness through the activation of TGF-β1. J Cancer. 2020;11:3803-15.
98. Mccarty JH. αvβ8 integrin adhesion and signaling pathways in development, physiology and disease. J Cell Sci. 2020;133:jcs239434.
100. Sasaki T, Timpl R. Domain IVa of laminin α5 chain is cell‐adhesive and binds β1 and αVβ3 integrins through Arg‐Gly‐Asp. FEBS Lett. 2001;509:181-5.
101. Munksgaard Thorén M, Chmielarska Masoumi K, Krona C, et al. Integrin α10, a novel therapeutic target in glioblastoma, regulates cell migration, proliferation, and survival. Cancers. 2019;11:587.
102. Ludwig BS, Kessler H, Kossatz S, Reuning U. RGD-binding integrins revisited: how recently discovered functions and novel synthetic ligands (Re-)shape an ever-evolving field. Cancers. 2021;13:1711.
103. Xiao T, Takagi J, Coller BS, Wang J, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59-67.
104. Tselepis VH, Green LJ, Humphries MJ. An RGD to LDV motif conversion within the disintegrin kistrin generates an integrin antagonist that retains potency but exhibits altered receptor specificity. J Biol Chem. 1997;272:21341-8.
105. Lafoya B, Munroe J, Miyamoto A, et al. Beyond the matrix: the many non-ECM ligands for integrins. Int J Mol Sci. 2018;19:449.
106. Hussein HAM, Walker LR, Abdel-Raouf UM, Desouky SA, Montasser AKM, Akula SM. Beyond RGD: virus interactions with integrins. Arch Virol. 2015;160:2669-81.
107. Davis PJ, Mousa SA, Cody V, Tang H, Lin H. Small molecule hormone or hormone-like ligands of integrin αVβ3: implications for cancer cell behavior. Horm Canc. 2013;4:335-42.
108. Kechagia JZ, Ivaska J, Roca-Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. 2019;20:457-73.
109. Moreno-layseca P, Icha J, Hamidi H, Ivaska J. Integrin trafficking in cells and tissues. Nat Cell Biol. 2019;21:122-32.
111. Boppart MD, Mahmassani ZS. Integrin signaling: linking mechanical stimulation to skeletal muscle hypertrophy. Am J Physiol Cell Physiol,. 2019;317:C629-41.
112. Lee J, Bankston LA, Robert C Liddington MAA. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure. 1995;3:1333-40.
113. Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288-300.
114. Rognoni E, Ruppert R, Fässler R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci. 2016;129:17-27.
115. Böttcher RT, Veelders M, Rombaut P, et al. Kindlin-2 recruits paxillin and Arp2/3 to promote membrane protrusions during initial cell spreading. J Cell Biol. 2017;216:3785-98.
116. Zhao X, Guan J. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Delivery Rev. 2011;63:610-5.
117. Murphy JM, Jeong K, Lim SS. FAK family kinases in vascular diseases. Int J Mol Sci. 2020;21:3630.
118. Calderwood DA, Fujioka Y, De Pereda JM, et al. Integrin β cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proc Natl Acad Sci USA. 2003;100:2272-7.
119. Kanter RK. Pediatric Emergency intravenous access: evaluation of a protocol. Am J Dis Child. 1986;140:132.
120. Meisaprow P, Aksorn N, Vinayanuwattikun C, Chanvorachote P, Sukprasansap M. Caffeine induces G0/G1 cell cycle arrest and inhibits migration through integrin αv, β3, and FAK/Akt/c-Myc signaling pathway. Molecules. 2021;26:7659.
121. Yuan R, Li Y, Yang B, et al. LOXL1 exerts oncogenesis and stimulates angiogenesis through the LOXL1-FBLN5/αvβ3 integrin/FAK-MAPK axis in ICC. Mol Ther Nucl Acids. 2021;23:797-810.
122. Sun W, Chan SY. Pulmonary arterial stiffness: an early and pervasive driver of pulmonary arterial hypertension. Front Med. 2018;5:204.
123. Chelladurai P, Seeger W, Pullamsetti SS. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur Respir J. 2012;40:766-82.
124. Hindmarch CCT, Tian L, Xiong PY, et al. An integrated proteomic and transcriptomic signature of the failing right ventricle in monocrotaline induced pulmonary arterial hypertension in male rats. Front Physiol. 2022;13:966454.
125. Meng L, Liu X, Teng X, et al. Osteopontin plays important roles in pulmonary arterial hypertension induced by systemic‐to‐pulmonary shunt. FASEB J. 2019;33:7236-51.
126. Blanchard N, Link PA, Farkas D, et al. Dichotomous role of integrin‐β5 in lung endothelial cells. Pulm Circ. 2022;12:e12156.
127. Sun L, Li H, Li Y, et al. Proteomic insights into platelet dysregulation and pathogenic mechanisms of chronic thromboembolic pulmonary hypertension. J Transl Med. 2025;23:1074.
128. Befani C, Liakos P. Hypoxia upregulates integrin gene expression in microvascular endothelial cells and promotes their migration and capillary‐like tube formation. Cell Biol Int. 2017;41:769-78.
129. Webb DJ, Donais K, Whitmore LA, et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6:154-61.
130. Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435-78.
131. Aikawa R, Nagai T, Kudoh S, et al. Integrins play a critical role in mechanical stress-induced p38 MAPK activation. Hypertension. 2002;39:233-8.
132. Wang R, Xu J, Wu J, Gao S, Wang Z. Angiotensin‑converting enzyme 2 alleviates pulmonary artery hypertension through inhibition of focal adhesion kinase expression. Exp Ther Med. 2021;22:1165.
133. Mehta D, Tiruppathi C, Sandoval R, Minshall RD, Holinstat M, Malik AB. Modulatory role of focal adhesion kinase in regulating human pulmonary arterial endothelial barrier function. J Physiol. 2002;539:779-89.
134. Paulin R, Meloche J, Courboulin A, et al. Targeting cell motility in pulmonary arterial hypertension. Eur Respir J. 2014;43:531-44.
135. Chen H, Liu P, Pan X, et al. Downregulation of collagen IV deposition and ITGB1-FAK signaling pathway to inhibit adipogenesis: a novel mechanism of swertiamarin in treating type 2 diabetes mellitus. Int J Biol Macromol. 2025;299:140048.
136. Niit M, Hoskin V, Carefoot E, et al. Cell-cell and cell-matrix adhesion in survival and metastasis: Stat3 versus Akt. Biomol Concepts. 2015;6:383-99.
137. Liu C, Chen K, Wang H, et al. Gastrin attenuates renal ischemia/reperfusion injury by a PI3K/Akt/Bad-mediated anti-apoptosis signaling. Front Pharmacol. 2020;11:540479.
139. Sun T, Zhang J, Deng B, et al. FOXO1 and FOXO3a sensitize non-small-cell lung cancer cells to cisplatin-induced apoptosis independent of Bim. Acta Biochim Biophys Sin. 2020;52:1348-59.
141. Abeyrathna P, Kovacs L, Han W, Su Y. Calpain-2 activates Akt via TGF-β1-mTORC2 pathway in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol. 2016;311:C24-34.
142. Fan Z, Li C, Qin C, et al. Role of the PI3K/AKT pathway in modulating cytoskeleton rearrangements and phenotype switching in rat pulmonary arterial vascular smooth muscle cells. DNA Cell Biol. 2014;33:12-9.
143. Gu C, Yang Z, Su S, et al. 4-Terpineol attenuates pulmonary vascular remodeling via suppressing PI3K/Akt signaling pathway in hypoxia-induced pulmonary hypertension rats. Toxicol Appl Pharmacol. 2023;473:116596.
144. Vandamme D, Herrero A, Al-Mulla F, Kolch W. Regulation of the MAPK pathway by raf kinase inhibitory protein. Crit Rev Oncog. 2014;19:405-15.
145. Liu Y, Tang BL, Lu ML, Wang HX. Astragaloside IV improves pulmonary arterial hypertension by increasing the expression of CCN1 and activating the ERK1/2 pathway. J Cell Mol Med. 2023;27:622-33.
146. Church AC, Martin DH, Wadsworth R, et al. The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK-alpha: a potential novel anti-inflammatory strategy in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2015;309:L333-47.
147. Das M, Zawada WM, West J, Stenmark KR. JNK2 regulates vascular remodeling in pulmonary hypertension. Pulm Circ. 2018;8:1-13.
148. Ding Q, Stewart J, Olman MA, Klobe MR, Gladson CL. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J Biol Chem. 2003;278:39882-91.
149. Ishigaki T, Imanaka‐yoshida K, Shimojo N, Matsushima S, Taki W, Yoshida T. Tenascin‐C enhances crosstalk signaling of integrin αvβ3/PDGFR‐β complex by SRC recruitment promoting PDGF‐induced proliferation and migration in smooth muscle cells. J Cell Physiol. 2011;226:2617-24.
150. Jia D, Zhu Q, Liu H, et al. Osteoprotegerin disruption attenuates HySu-induced pulmonary hypertension through integrin αvβ3/FAK/AKT pathway suppression. Circ Cardiovasc Genet. 2017;10:e001591.
151. Kökény G, Calvier L, Hansmann G. PPARγ and TGFβ—Major regulators of metabolism, inflammation, and fibrosis in the lungs and kidneys. Int J Mol Sci. 2021;22:10431.
152. Gorelova A, Berman M, Al Ghouleh I. Endothelial-to-mesenchymal transition in pulmonary arterial hypertension. Antioxid Redox Signal. 2021;34:891-914.
153. Bai P, Lyu L, Yu T, et al. Macrophage-derived legumain promotes pulmonary hypertension by activating the MMP (matrix metalloproteinase)-2/TGF (transforming growth factor)-β1 signaling. Arterioscler Thromb Vasc Biol. 2019;39:e130-45.
154. Rifkin D, Sachan N, Singh K, Sauber E, Tellides G, Ramirez F. The role of LTBPs in TGF beta signaling. Dev Dyn. 2021;251:75-84.
155. Munger JS, Huang X, Kawakatsu H, et al. A mechanism for regulating pulmonary inflammation and fibrosis: the integrin αvβ6 binds and activates latent TGF β1. Cell. 1999;96:319-28.
156. Margadant C, Sonnenberg A. Integrin-TGF‐β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97-105.
157. Andreucci E, Bugatti K, Peppicelli S, et al. Nintedanib-αVβ6 integrin ligand conjugates reduce TGFβ-induced EMT in human non-small cell lung cancer. Int J Mol Sci. 2023;24:1475.
158. Decaris ML, Schaub JR, Chen C, et al. Dual inhibition of αvβ6 and αvβ1 reduces fibrogenesis in lung tissue explants from patients with IPF. Respir Res. 2021;22:265.
159. Gao C, Li J, Feng F, et al. EDIL3 regulates pulmonary artery smooth muscle cell proliferation and migration via integrin αVβ3/ERK1/2 axis in pulmonary hypertension. Eur J Pharmacol. 2025;1003:177901.
160. Lafyatis R. Transforming growth factor β—at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10:706-19.
161. Mitra A, Agarwal S, Chakraborty A, et al. Loss of ROR2 tyrosine kinase receptor is associated with endothelial dysfunction in PAH via inappropriate integrin β1 activation. Hypertension. 2026;83.
162. Qi R, Zhang Y, Yan F. Exosomes enriched by miR-429-3p derived from ITGB1 modified Telocytes alleviates hypoxia-induced pulmonary arterial hypertension through regulating Rac1 expression. Cell Biol Toxicol. 2024;40:32.
163. Hoeper MM, Badesch DB, Ghofrani HA, et al. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med. 2023;388:1478-90.
164. Ricart AD, Tolcher AW, Liu G, et al. Volociximab, a chimeric monoclonal antibody that specifically binds α5β1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin Cancer Res. 2008;14:7924-9.
165. Cianfrocca ME, Kimmel KA, Gallo J, et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2), a beta integrin antagonist, in patients with solid tumours. Br J Cancer. 2006;94:1621-6.
166. Yurdagul A, Green J, Albert P, Mcinnis MC, Mazar AP, Orr AW. α5β1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1362-73.
167. Vansteenkiste J, Barlesi F, Waller C, et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: results of an open-label, randomized, controlled phase II study (CERTO). Ann Oncol. 2015;26:1734-40.
168. Macdonald TJ, Vezina G, Stewart CF, et al. Phase II study of cilengitide in the treatment of refractory or relapsed high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol. 2013;15:1438-44.
169. Reardon DA, Fink KL, Mikkelsen T, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610-7.
170. Vermorken J, Peyrade F, Krauss J, et al. Cisplatin, 5-fluorouracil, and cetuximab (PFE) with or without cilengitide in recurrent/metastatic squamous cell carcinoma of the head and neck: results of the randomized phase I/II ADVANTAGE trial (phase II part). Ann Oncol. 2014;25:682-8.
171. Zhou X, Zhang J, Haimbach R, et al. An integrin antagonist (MK‐0429) decreases proteinuria and renal fibrosis in the ZSF1 rat diabetic nephropathy model. Pharmaco Res Perspec. 2017;5:e00354.
172. Zhang J, Wang T, Saigal A, et al. Discovery of a new class of integrin antibodies for fibrosis. Sci Rep. 2021;11:2118.
173. John AE, Graves RH, Pun KT, et al. Translational pharmacology of an inhaled small molecule αvβ6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat Commun. 2020;11:4659.
174. Raghu G, Mouded M, Chambers DC, et al. A phase IIb randomized clinical study of an anti-αvβ6 monoclonal antibody in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2022;206:1128-39.
175. Mooney JJ, Jacobs S, Lefebvre ÉA, et al. Bexotegrast shows dose-dependent integrin αvβ6 receptor occupancy in lungs of participants with idiopathic pulmonary fibrosis: a phase 2, open-label clinical trial. Ann Am Thorac Soc. 2025;22:350-8.
176. Mu Y, Liu J, Wu Q, et al. A dual αvβ1/αvβ6 integrin inhibitor Bexotegrast (PLN-74809) ameliorates organ injury and fibrogenesis in fibrotic kidney disease. Eur J Pharmacol. 2024;983:176983.
177. Slack RJ, Macdonald SJF, Roper JA, Jenkins RG, Hatley RJD. Emerging therapeutic opportunities for integrin inhibitors. Nat Rev Drug Discov. 2021;21:60-78.
178. Li Z, Zhou Y, Ding Y, Guo Q, Zhao L. Roles of integrin in tumor development and the target inhibitors. Chin J Nat Med. 2019;17:241-51.
179. Stupp R, Hegi ME, Gorlia T, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1100-8.
180. Agrawal V, Hemnes AR. Integrin targeting therapies in pulmonary arterial hypertension: a roadmap for traversing the translational valley of death? Circulation. 2025;151:1184-6.
181. Zhang S, Zhang Q, Lu Y, et al. Roles of integrin in cardiovascular diseases: from basic research to clinical implications. Int J Mol Sci. 2024;25:4096.
182. Simone E, Ding B, Muzykantov V. Targeted delivery of therapeutics to endothelium. Cell Tissue Res. 2008;335:283-300.
183. Zhou X, Liu H, Zhao H, Wang T. RGD-modified nanoliposomes containing quercetin for lung cancer targeted treatment. Onco Targets Ther. 2018;11:5397-405.
184. Castro Brás LE, Frangogiannis NG. Extracellular matrix-derived peptides in tissue remodeling and fibrosis. Matrix Biol. 2020;91-92:176-87.
185. Nikolov A, Popovski N. Extracellular matrix in heart disease: focus on circulating collagen type I and III derived peptides as biomarkers of myocardial fibrosis and their potential in the prognosis of heart failure: a concise review. Metabolites. 2022;12:297.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].