


Panvascular aging as a unifying framework: convergent mechanisms across age-related diseases
 0
0 
Abstract
Panvascular aging is characterized by a systemic decline in vascular integrity that underlies the pathogenesis of multiple age-related diseases. Rather than affecting isolated vascular beds, aging triggers widespread changes across the entire circulatory system, including both large vessels and microcirculation in multiple organs. Core mechanisms encompass endothelial dysfunction, glycocalyx degradation, oxidative stress, inflammaging, and disturbed mechanotransduction, which converge to impair vascular homeostasis. At the cellular level, telomere shortening, epigenetic remodeling, and vascular smooth muscle cell phenotypic switching accelerate senescence and extracellular matrix stiffening. Importantly, panvascular aging propagates through systemic inter-organ axes, such as the brain-vascular and heart-kidney networks, thereby amplifying shared pathogenic pathways and promoting organ dysfunction. Consequently, it constitutes an integrative framework linking cardiovascular disease, diabetes, chronic kidney disease, and neurodegeneration. Recognizing panvascular aging as a common pathophysiological axis underscores the importance of developing systemic strategies that enhance vascular resilience beyond singular disease-oriented approaches. Emerging therapeutic strategies - including senotherapeutics, epigenetic modulators, glycocalyx restoration, and metabolic restoration - are being explored as promising interventions to attenuate vascular aging and mitigate multimorbidity. This review synthesizes current insights into the molecular and mechanobiological mechanisms of panvascular aging and frames it as a conceptual keystone in aging biology, highlighting translational opportunities for the prevention and treatment of chronic age-related diseases.
Keywords
INTRODUCTION
Defining panvascular aging
Panvascular aging refers to the progressive, systemic degeneration of the entire vascular network, including arteries, veins, and capillaries which are driven by shared molecular and cellular hallmarks of aging. Unlike localized vascular pathology, it reflects coordinated alterations across macrovascular and microvascular beds, leading to multi-organ dysfunction. This concept underscores that panvascular aging is not merely aging occurring in multiple vessels, but rather an integrative biological program that links systemic aging processes with vascular heterogeneity[1]. Key mechanisms such as endothelial dysfunction, chronic low-grade inflammation, oxidative stress, mitochondrial impairment, and vascular smooth muscle cells (VSMCs) remodeling progressively impair vasomotor responses, increase vascular stiffness, and disrupt microcirculatory regulation, thereby predisposing multiple organ systems to age-related decline[2].
Systemic nature and clinical significance
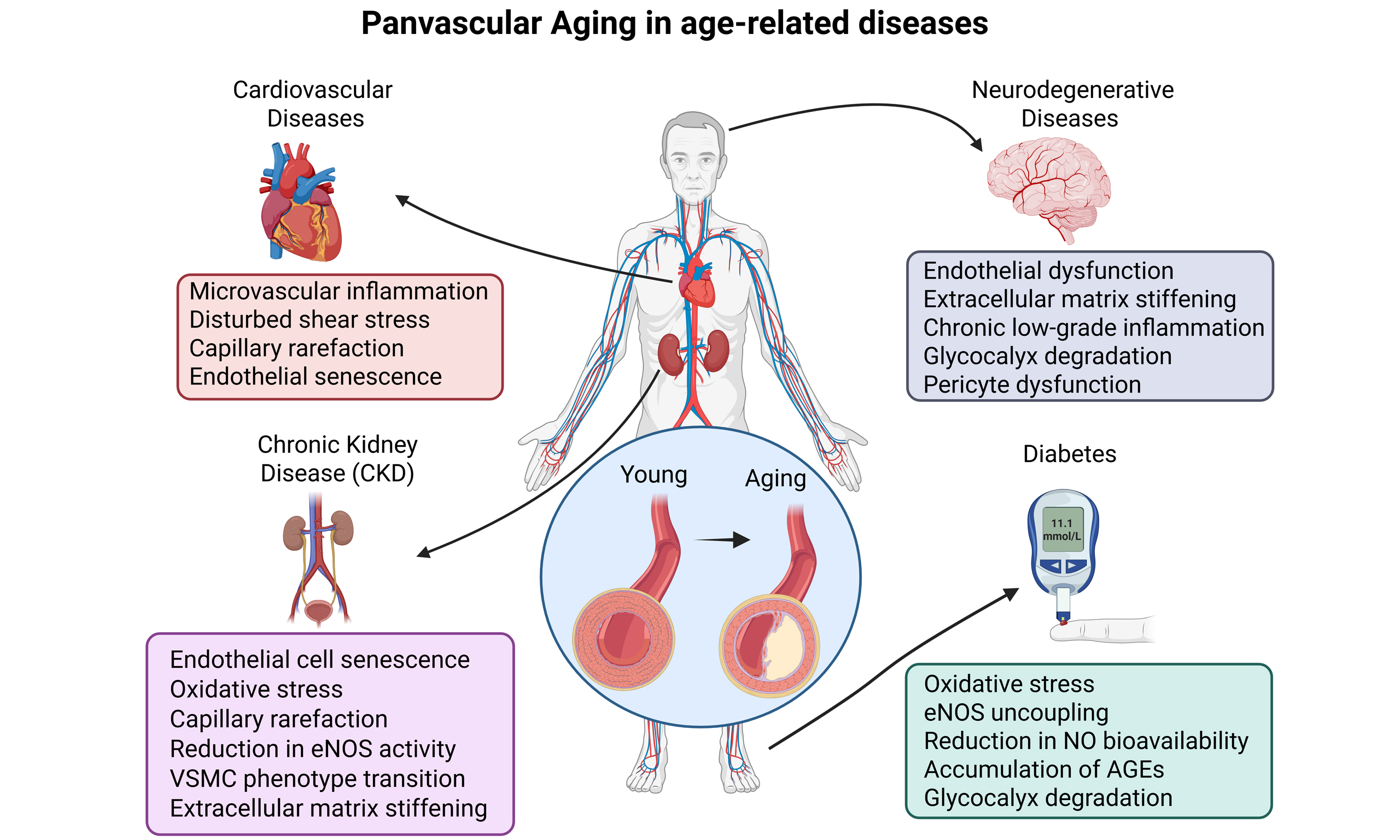
The systemic nature of panvascular aging positions it as a unifying pathogenic driver across diverse chronic diseases. Cardiovascular and cerebrovascular disorders represent prominent clinical manifestations, while neurodegenerative diseases[3], chronic kidney disease (CKD)[4], and metabolic syndromes such as diabetes[5] similarly exemplify conditions in which vascular aging functions not merely as a consequence, but as a central pathogenic determinant. For instance, elastin fragmentation and collagen cross-linking in large arteries elevate pulse wave velocity and systolic load, contributing to heart failure and stroke risk[6,7]. Concurrently, endothelial senescence and capillary rarefaction impair cerebral perfusion and clearance mechanisms, accelerating cognitive decline[8]. Likewise, renal microvascular stiffening and glomerular endothelial dysfunction exacerbate nephron loss, while insulin resistance and hyperglycemia intensify oxidative stress, further perpetuating systemic vascular aging[9]. Thus, panvascular aging acts as both a common denominator and a pathophysiological amplifier across organ systems[10].
Distinction from related concepts
Panvascular aging differs from general vascular aging[11] - which primarily describes age-related remodeling of large arteries - and from endothelial aging[12], which is confined to cellular deterioration of endothelial cells (ECs). Instead, panvascular aging provides a broader, mechanistic framework that integrates aging phenotypes across all vascular beds and cell types, explicitly linking vascular aging to multi-organ dysfunction [Figure 1].
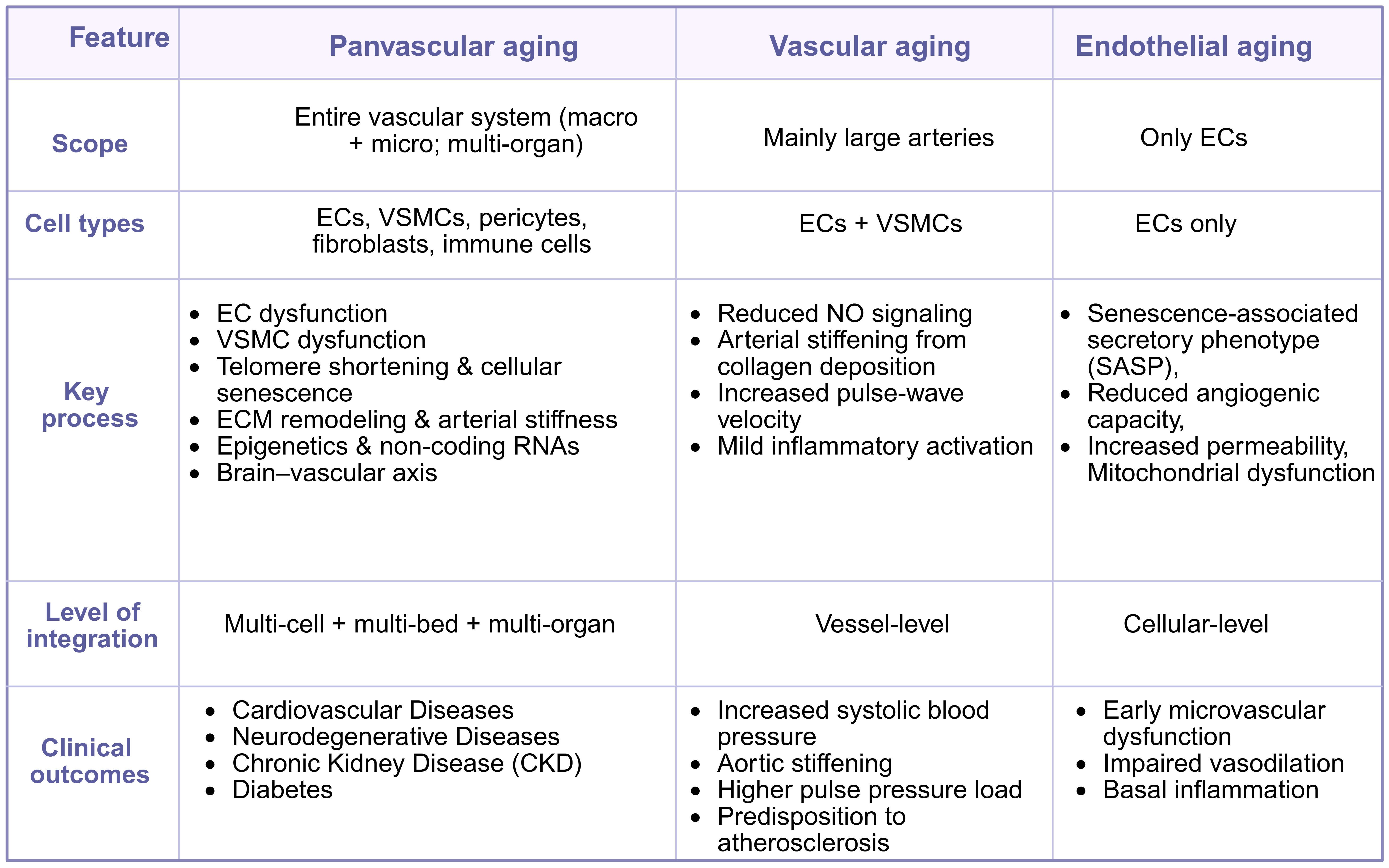
Figure 1. Comparative overview of vascular aging phenotypes. This table summarizes the defining features of three interrelated aging processes within the vasculature: Panvascular aging, Vascular aging, and Endothelial aging. The figure was created with https://app.biorender.com/citation/69396cbaf8ae54942d60cecc. ECs: Endothelial cells; VSMCs: vascular smooth muscle cells; NO: nitric oxide.
Evidence for systemically synchronized vascular aging
High-dimensional omics data offer compelling support for the coordinated nature of vascular aging. Large-scale plasma proteomic analyses reveal age-dependent protein signatures that correlate with the biological aging of multiple organs simultaneously[13]. These circulating signatures are enriched for vascular pathways - including insulin-like growth factor (IGF)/insulin signaling[14], extracellular matrix (ECM) turnover[14], complement activation, and endothelial inflammatory mediators - supporting the existence of a systemic axis that integrates vascular aging trajectories across tissues. Similarly, cross-tissue single-cell atlases in mice have identified conserved endothelial senescence programs[15], marked by upregulation of Intercellular Adhesion Molecule 1 (ICAM1), Serpin Family E Member 1 (SERPINE1), and pro-inflammatory senescence-associated secretory phenotype (SASP) components[16], that appear synchronously in diverse vascular beds with advancing age.
Experimental evidence for circulating regulators
Parabiosis and plasma-transfer studies provide causal evidence that vascular aging is modulated by blood-borne factors. Heterochronic parabiosis models demonstrate that a young systemic milieu can partially reverse vascular dysfunction in aged partners. For example, exposure to young blood has been shown to restore cerebromicrovascular endothelial nitric oxide (NO) bioavailability, improve neurovascular coupling, and reduce oxidative stress in aged mice[17]. Conversely, old blood accelerated endothelial senescence and impaired vascular reactivity in young counterparts[18,19]. Complementary plasma-transfer studies indicate that young plasma or its small extracellular vesicles can rejuvenate microvascular density, mitochondrial function, and endothelial barrier integrity in aged tissues[20]. These findings collectively support a model in which diffusible endocrine, immunologic, or vesicle-mediated factors orchestrate vascular aging at the organismal level.
Clinical corroboration of systemic coordination
Clinical observations further reinforce the systemic view, demonstrating coherence of vascular aging biomarkers across organs. Measures of macrovascular aging such as carotid-femoral pulse wave velocity (cfPWV) correlate strongly with microvascular dysfunction in the retina, kidney, and brain, independent of traditional risk factors. For instance, increased aortic stiffness predicts retinal arteriolar narrowing, reduced cerebral perfusion, and early CKD, indicating a shared upstream process of systemic vascular stiffening[6]. Endothelial function metrics, such as flow-mediated dilation (FMD), also correlate with cerebral small-vessel disease burden, renal microalbuminuria, and peripheral microvascular reactivity, suggesting coordinated multi-organ endothelial aging[21]. Longitudinal cohorts further reveal that vascular aging trajectories in one organ system frequently predict aging phenotypes in other vascular beds, consistent with a systemic pathophysiology[22].
Mechanistic coherence across vascular beds
At the mechanistic level, common hallmarks - including mitochondrial dysfunction[2], impaired NO signaling[23], endothelial senescence, chronic inflammation, and maladaptive ECM remodeling[24] - are shared across vascular beds and appear synchronized by systemic modulators such as circulating cytokines [e.g., interleukin (IL)-6, tumor necrosis factor (TNF)-α][25], metabolic signals [e.g., advanced glycation end-products (AGEs), lipids][26,27], and endocrine regulators [e.g., Insulin-like growth factor 1 (IGF1), Klotho][28,29]. Importantly, interventions targeting these systemic pathways exert benefits across multiple vascular territories rather than in isolated beds[30], highlighting the integrated nature of panvascular aging.
Implications for biomarkers and therapeutics
Recognizing panvascular aging as a coordinated systemic phenomenon has important translational implications[22]. Biomarkers of vascular aging should be multi-organ and circulation-based, while therapeutic strategies targeting the systemic milieu - such as senolytics, anti-inflammatory agents, nicotinamide adenine dinucleotide (NAD+) boosters, or circulatory-factor modulation - may yield broad, panvascular benefits across the lifespan[2].
This review synthesizes current understanding of the shared mechanisms driving panvascular aging and their role in the pathogenesis of major age-related diseases. Focusing on endothelial dysfunction, oxidative stress, inflammatory signaling, and VSMCs abnormalities as central hubs, we propose an integrated framework to elucidate how panvascular aging coordinates systemic disease progression. By integrating mechanistic insights with translational perspectives, we highlight the necessity of comprehensive strategies to maintain vascular health and extend healthspan in aging populations.
PANVASCULAR AGING AS A HETEROGENEOUS, MULTI-SCALE PROCESS
“Panvascular aging” refers to the integrated effects of aging on the entire vascular system, yet it is increasingly understood as a heterogeneous, rather than uniform, process[2]. The manifestations and mechanisms of aging differ considerably depending on the vascular bed[31] - such as large elastic arteries versus small resistance arteries versus organ-specific microvessels - and on the cell type involved[32], including ECs, VSMCs, and pericytes or mural cells. Advances in single-cell and spatial transcriptomics are now beginning to illuminate the complexity underlying this heterogeneity.
Macro- versus microvascular beds: distinct aging signatures
In large elastic arteries such as the ascending thoracic aorta, chronological aging typically leads to increased arterial stiffness, reduced distensibility, and structural remodeling of the vessel wall. A recent single-cell transcriptomic study of the aged ascending thoracic aorta, for example, revealed increased collagen deposition in the medial and adventitial layers, decreased abundance of VSMCs, enhanced inflammatory signaling, and T-lymphocyte enrichment in the adventitia compared with young controls[33,34]. These molecular and cellular alterations corresponded functionally to a decline in passive cyclic distensibility.
In contrast, aging in resistance arteries and smaller muscular arteries is characterized more prominently by VSMC phenotypic switching (from a contractile to a synthetic or senescent state), ECM remodeling, and altered vasoreactivity - processes that differ from the stiffening-dominant profile seen in large elastic vessels. A study of aged hypertensive and normotensive rats, using single-cell RNA sequencing of aorta, femoral, and mesenteric arteries, demonstrated age- and hypertension-associated shifts in VSMCs composition: contractile VSMCs declined, while synthetic, apoptotic, and senescent subpopulations expanded[35].
At the microvascular level - for instance, in capillaries of the brain, adipose tissue, retina, and kidney - aging manifests as capillary rarefaction (loss of density), impaired angiogenesis, endothelial dysfunction, basement-membrane thickening, altered pericyte coverage, and disruption of blood-tissue barriers. Accumulating evidence highlights a central role for endothelial senescence in the age-associated deterioration of the blood-brain barrier (BBB) and blood-retinal barrier (BRB)[36].
Moreover, aging in subcutaneous adipose tissue is associated with a reduced proportion of venular ECs and a pseudotemporal shift toward a terminal metabolic state marked by suppressed oxidative phosphorylation and enhanced inflammatory signaling, with C-X-C motif chemokine ligand 9 (CXCL9) identified as a potential mediator linking endothelial metabolic dysfunction to impaired angiogenesis[37]. These findings highlight the heterogeneity of microvascular aging across vascular beds and endothelial subpopulations.
Thus, the dominant aging processes vary by vascular compartment: large elastic arteries undergo stiffening and ECM remodeling; muscular and resistance arteries exhibit VSMC phenotypic switching and functional remodeling; and microvessels display capillary rarefaction, barrier impairment, and ECs/pericyte dysfunction.
Cellular heterogeneity: ECs, VSMCs, and pericytes
ECs
As the inner lining of all vessels, ECs are directly exposed to circulating factors and hemodynamic forces, playing central roles in vasomotor regulation, barrier function, angiogenesis, and leukocyte adhesion. Accumulating evidence indicates that ECs in different tissues do not age identically: although common “core” aging-associated features - such as increased inflammatory gene expression, senescence-related signatures, and a reduced proportion of capillary ECs - are observed, tissue-specific aging patterns also emerge, affecting angiogenic capacity, barrier integrity, DNA-repair pathways, and extracellular-matrix regulation[38]. Single-cell transcriptomic analyses of cardiac ECs from young and aged mice have further identified multiple EC subclusters and revealed pronounced age-associated shifts in both their cellular composition and transcriptional programs, indicating that certain EC subtypes are disproportionately vulnerable to aging[39]. These findings support the concept that endothelial aging is intrinsically heterogeneous, with some EC subpopulations retaining youthful characteristics for longer periods, while others preferentially transition toward senescent or dysfunctional states.
VSMCs and large-vessel remodeling
VSMCs, the main mural cells in medium- to large-sized arteries, provide contractile force and contribute to vessel-wall stability. Aging, often in conjunction with hypertension, drives VSMC phenotypic switching from a contractile to a synthetic, proliferative, ECM-producing, senescent, or even apoptotic phenotype. The hypertensive-aging single-cell study noted an increased abundance of synthetic/senescent VSMCs in aged arteries[35]. Bulk transcriptomic analyses of aging aortic tissues further reveal dysregulation not only of classical senescence-associated genes, but also of pathways such as molecular chaperones [e.g., 70-kDa heat shock protein (HSP70) family] and circadian clock genes, indicating broader disruptions in proteostasis and circadian regulation in the aged arterial wall[40].
These molecular alterations likely contribute to ECM remodeling, collagen accumulation, elastin fragmentation, and the resultant increases in stiffness and loss of compliance. Consequently, interventions targeting vascular aging - such as senolytics or ECM-directed agents - may need to address VSMC phenotype and ECM homeostasis in large arteries, but are unlikely to suffice for microvascular or endothelial-specific pathologies.
Pericytes and mural cells in the microvasculature
Pericytes and other mural cells are critical for capillary stability, blood-flow regulation, and blood-tissue barrier integrity, particularly in microvascular beds of the brain, retina, adipose tissue, and kidney. Recent studies highlight pericyte dysfunction as a key contributor to microvascular aging. Pericytes from aged rat brains display senescence-associated phenotypes and diminished support of BBB function[41], while in vivo studies in aging mice show that pericyte loss promotes capillary dilation, flow heterogeneity, stalling and regression, defective remodeling, and reduced capillary network connectivity[42].
Pericytes are increasingly recognized as a heterogeneous population, varying by tissue origin[43], developmental lineage[44], and local microenvironment. Their functions in vessel stabilization, angiogenesis, and tissue homeostasis are shaped by distinct signaling pathways, including Platelet-derived growth factor β/Platelet-derived growth factor receptor β (PDGF-β/PDGFRβ), Transforming growth factor-β (TGF-β), Notch, Vascular endothelial growth factor (VEGF), and Mechanistic target of rapamycin/Transcription factor forkhead box O (mTOR/FOXO)[45-48]. During aging, these regulatory pathways may become dysregulated in a tissue- or context-specific manner, leading to selective loss of certain pericyte subtypes and consequently to capillary rarefaction or barrier breakdown in particular organs (e.g., brain, retina, kidney). Loss of pericyte coverage and basement-membrane thickening, observed in aged microvessels, can weaken EC-pericyte contacts, destabilize capillaries, and impair microvascular perfusion[42,49].
Notably, age-associated venular EC loss and microvascular rarefaction in adipose tissue are accompanied by metabolic reprogramming of aged ECs, marked by suppressed oxidative phosphorylation and increased CXCL9 expression, implicating coordinated inflammatory-metabolic dysregulation in microvascular aging[37]. Together, these findings indicate that pericytes are active determinants of microvascular aging, and their heterogeneity across organs likely underlies the differential vulnerability of microvascular beds (e.g., brain vs. kidney vs. retina vs. adipose) to aging-associated dysfunction.
ENDOTHELIAL DYSFUNCTION
Decline of nitric oxide bioavailability
With advancing age, endothelial NO bioavailability declines throughout the vascular system due to multiple, interconnected mechanisms that collectively accelerate panvascular aging. Endothelial NO synthase (eNOS), the principal enzymatic source of vascular NO, becomes “uncoupled” under conditions of oxidative stress and tetrahydrobiopterin (BH4) depletion, resulting in superoxide production rather than NO and thereby exacerbating vascular redox imbalance and dysfunction[23,50]. Replicative endothelial senescence further contributes by elevating the BH2/BH4 ratio and downregulating eNOS expression, shifting enzymatic output toward reactive oxygen species (ROS)[51]. Clinical studies in healthy adults demonstrate that aging significantly increases circulating BH2 without reducing BH4, yet still impairs microvascular function, as evidenced by diminished cutaneous blood flow[52]. Animal models reinforce these findings: endothelial-specific deletion of guanosine triphosphate cyclohydrolase 1 (GCH1) sensitizes the vasculature to angiotensin II-induced remodeling, hypertension, and aneurysm formation, underscoring the pathological role of BH4 insufficiency[53]. Notably, acute BH4 supplementation in older adults fails to improve FMD, suggesting that cofactor replacement alone is insufficient in the context of established endothelial senescence[54].
Emerging evidence identifies adenosine monophosphate (AMP)-activated protein kinase (AMPK) as a pivotal upstream regulator of the BH4-eNOS axis. AMPK activation stabilizes GCH1, enhances BH4 synthesis, and promotes eNOS recoupling. For example, intermedin (IMD) protects the renal microcirculation by activating the AMPK-GCH1-BH4 pathway, thereby preventing eNOS uncoupling and attenuating CKD progression[55]. Similarly, the adipokine C1q/TNF-related protein 13 (CTRP13) ameliorates endothelial ferroptosis and dysfunction through AMPK-GCH1-BH4 signaling[56]. Pharmacological AMPK activators, including metformin and Glucagon-like peptide-1 (GLP-1) receptor agonists, also restore NO signaling via this pathway, positioning metabolic modulation as a therapeutic frontier[57].
Chronic inflammation further impairs NO bioavailability in panvascular aging. Elevated levels of the chemokine CCL4 promote oxidative stress, impair angiogenesis, and suppress endothelial progenitor cell function in aged models, thereby worsening vascular rarefaction and dysfunction[58]. Neutralizing CCL4 restores NO signaling and mitigates endothelial injury, indicating that inflammatory chemokine blockade may synergize with BH4-targeted strategies to maintain vascular homeostasis[58]. This inflammatory amplification loop not only accelerates NO decline but also links systemic immune aging to endothelial senescence across vascular beds.
Beyond conduit arteries, NO insufficiency extends to microvascular territories, contributing to impaired neurovascular coupling, BBB disruption, and renal microvascular fragility in aging[59,60]. Gut microbiota dysbiosis further exacerbates systemic oxidative and inflammatory burden, indirectly impairing the BH4/NO pathway[60]. Recent insights also highlight that endothelial-derived hyperpolarizing factors (EDHFs), including epoxyeicosatrienoic acids (EETs) and hydrogen peroxide (H2O2), may partially compensate for diminished NO bioavailability in the microcirculation[61].
In summary, the interplay of BH4 depletion, eNOS uncoupling, chronic inflammation (including CCL4 signaling), and oxidative stress collectively erodes NO bioavailability throughout the vascular tree, representing a central mechanistic pillar of panvascular aging. Therapeutic strategies combining cofactor repletion, AMPK activation, anti-inflammatory modulation, and microbiome-targeted interventions offer promise for restoring vascular integrity and delaying the systemic progression of vascular aging [Figure 2].
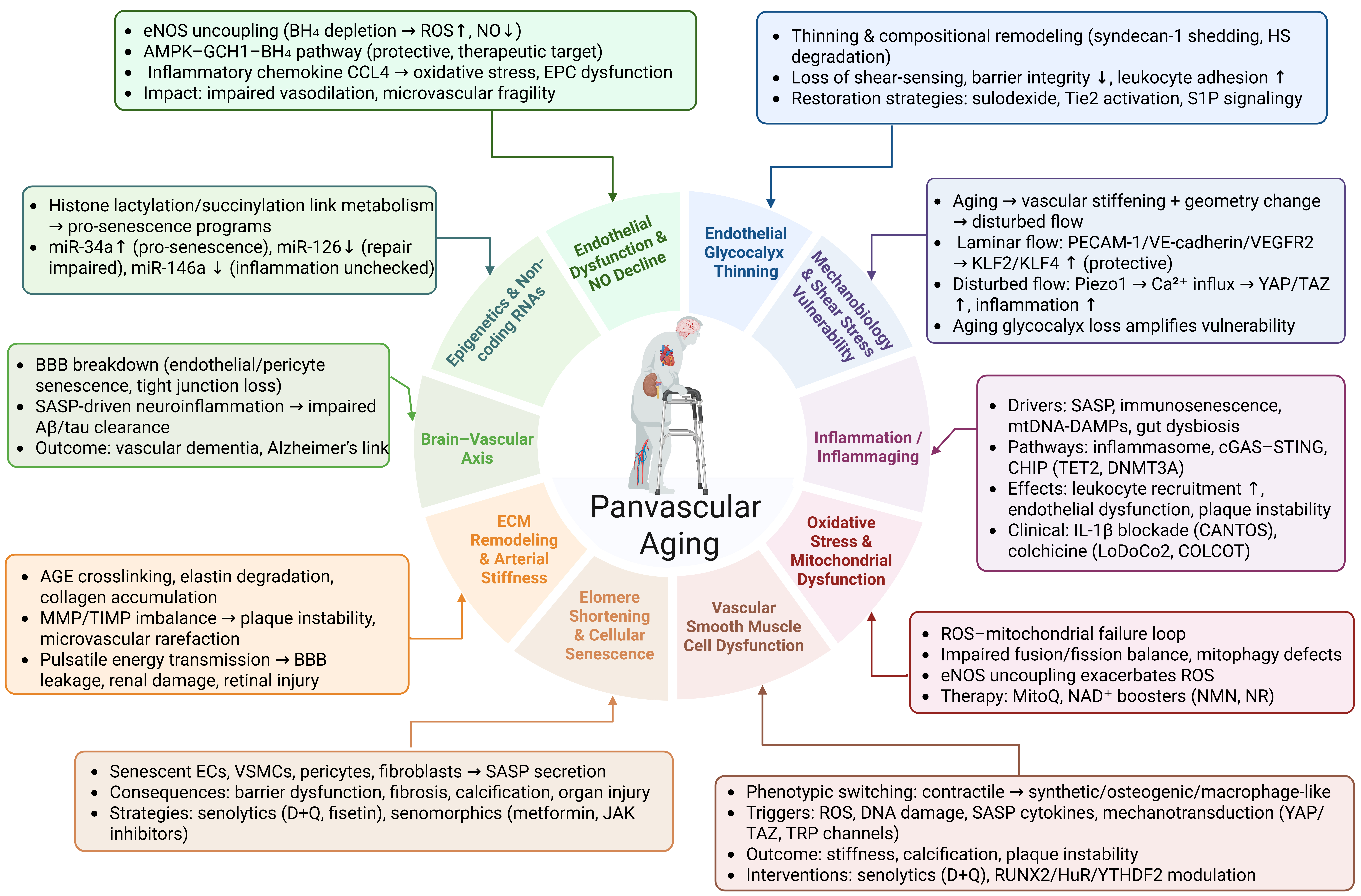
Figure 2. Integrated mechanisms of panvascular aging. Multiple interdependent processes - including NO decline, glycocalyx thinning, shear stress vulnerability, chronic inflammation, oxidative stress, VSMCs dysfunction, senescence, ECM remodeling, epigenetic reprogramming, and brain-vascular axis disruption - converge to accelerate systemic vascular deterioration and organ injury. Therapeutic strategies targeting BH4/NO signaling, AMPK activation, glycocalyx restoration, mechanotransduction reprogramming, anti-inflammatory interventions, mitochondrial protection, senotherapies, ECM modulation, and epigenetic/microRNA pathways hold promise for mitigating panvascular aging. The figure was created with https://app.biorender.com/citation/68c8320f7eaabe5720f1b2d6. ECs: Endothelial cells; VSMCs: vascular smooth muscle cells; NO: nitric oxide; AMPK: adenosine monophosphate-activated protein kinase; ECM: extracellular matrix; ROS: reactive oxygen species; eNOS: endothelial NO synthase; GCH1: guanosine triphosphate cyclohydrolase 1; BBB: blood-brain barrier; SASP: senescence-associated secretory phenotype; MMPs: matrix metalloproteinases; AGEs: advanced glycation end-products; TIMPs: tissue inhibitor of metalloproteinases; YAP: yes-associated protein; TAZ: transcriptional coactivator with PDZ-binding motif; NMN: nicotinamide mononucleotide; NR: nicotinamide riboside; KLF4/2: Krüppel-like factor4/2; CHIP: clonal hematopoiesis of indeterminate potential; LoDoCo2: Low-dose colchicine 2; COLCOT: colchicine cardiovascular outcomes trial; IL-1β: interleukin-1 beta; CANTOS: canakinumab anti-inflammatory thrombosis outcomes study; EPC: endothelial progenitor cell; HS: heparan sulfate; JAK: Janus kinase; cGAS-STING: cyclic GMP-AMP synthase-stimulator of interferon genes; PECAM1: platelet and endothelial cell adhesion molecule 1; VEGFR2: vascular endothelial growth factor receptor 2; TRP channels: transient receptor potential channels; RUNX2: RUNX family transcription factor 2; HuR: human antigen R; YTHDF2: YTH N6-methyladenosine RNA binding protein F2.
Endothelial glycocalyx thinning and restoration
In parallel with the well-characterized age-dependent decline in eNOS function, compelling evidence indicates that the endothelial glycocalyx (EG) - a carbohydrate‐rich luminal layer composed of proteoglycans, glycoproteins, and glycosaminoglycans (GAGs) - undergoes structural degeneration during panvascular aging. The EG plays an essential role in shear‐stress sensing, vascular permeability regulation, and NO bioavailability. With advancing age, the EG exhibits both thinning and compositional remodeling, including loss or altered expression of core proteins and changes in GAG side chains, which collectively compromise its functional capacity. This degradation disrupts mechanotransduction, reduces NO bioavailability, increases endothelial permeability, and exposes adhesion receptors that promote leukocyte and platelet recruitment, thereby converting a local vascular insult into a systemic pro-inflammatory and pro-coagulant state[62,63]. Consequently, EG deterioration acts as both a marker and a mechanistic driver of endothelial dysfunction, accelerating endothelial senescence and systemic vascular aging.
Recent studies have delineated the mechanisms underlying EG degradation and highlighted potential restorative interventions. In brain capillaries of aged mice, pronounced EG thinning is observed, accompanied by transcriptional remodeling of genes involved in mucin-type O-glycan biosynthesis and heparan sulfate metabolism[62]. Mechanistically, excessive ROS, pro-inflammatory cytokines (e.g., IL-6, TNF-α), and activation of sheddases such as heparanase and matrix metalloproteinases (MMPs) promote cleavage of proteoglycan ectodomains (notably syndecan-1) and GAGs. Clinical and translational observations further report elevated plasma levels of EG-shedding markers (e.g., syndecan-1 and heparan sulfate fragments) that correlate with arterial stiffness and endothelial dysfunction[63]. These circulating glycocalyx fragments act as damage-associated molecular patterns (DAMPs), which are capable of entering the circulation and propagating inflammation and endothelial activation at distant vascular beds. Moreover, endothelium-derived mediators released during glycocalyx loss - including angiopoietin-2, cytokines, and pro-coagulant factors, can form feed-forward regulatory loops that reinforce remote endothelial dysfunction and organ injury, positioning the EG as both a sentinel and a therapeutic target for interrupting maladaptive inter-organ signaling.
Restoration of EG structure is increasingly recognized as a pivotal anti-vascular-aging strategy. Sulodexide, for example, prevents syndecan-1 shedding, restores glycocalyx integrity, and improves vascular reactivity in experimental and early clinical settings[64,65]. Modulation of the Angiopoietin/Tie2 axis has also emerged as a promising approach: pharmacological Tie2 activation with AKB-9778 (Razuprotafib) increases EG thickness, reduces leukocyte adhesion, and attenuates inflammation[66,67]. Recent studies indicate that Tie2 modulation can reverse age-related endothelial dysfunction in specialized vascular beds[67]. In parallel, sphingosine-1-phosphate (S1P) signaling has been identified as a glycocalyx-stabilizing pathway that inhibits syndecan-1 shedding and suppresses inflammatory endothelial activation[68].
Together, these findings underscore that EG restoration is not merely a local repair process but a central strategy for counteracting endothelial senescence, preserving flow-mediated vasodilation, suppressing thrombo-inflammation, and thereby mitigating panvascular aging [Figure 2].
Mechanobiology and shear stress vulnerability
Recent mechanobiological investigations have demonstrated that age-related changes in vascular geometry, arterial wall stiffness, and EG degradation collectively reshape local hemodynamic profiles. These alterations increase the prevalence of disturbed or oscillatory shear stress across the arterial tree, accelerating panvascular aging and elevating susceptibility to atherothrombotic events[69,70].
Under physiological unidirectional (laminar) shear stress, ECs activate a junctional mechanosensory complex composed of Platelet And Endothelial Cell Adhesion Molecule 1 (PECAM-1), vascular endothelial cadherin (VE-cadherin), and Vascular endothelial growth factor receptor 2 (VEGFR2). Engagement of this complex initiates intracellular kinase signaling cascades, including the Mitogen-activated protein kinase 5-Extracellular Signal-Regulated Protein Kinase 5 (MEK5-ERK5) pathway, which upregulate the shear-responsive transcription factors Krüppel-like factor (KLF)2 and KLF4. These transcriptional programs maintain an endothelial phenotype characterized by anti-inflammatory, antioxidant, and NO-enriched properties, thereby preserving vascular homeostasis[69,71]. KLF2 and KLF4 induction constitutes a pivotal regulatory axis of flow-mediated vascular homeostasis, promoting eNOS expression and genes with anticoagulant and anti-adhesive functions, while suppressing nuclear factor-kappa B (NF-κB)-dependent inflammatory signaling. However, this protective endothelial program progressively declines with aging and in vascular regions exposed to disturbed flow[72,73].
Conversely, disturbed shear stress activates mechanosensitive ion channels and transcriptional co-activators that reprogram ECs toward a pro-inflammatory state. The mechanically gated ion channel Piezo1 serves as a critical upstream sensor; under disturbed flow, it mediates sustained Ca2+ influx that propagates pro-inflammatory signaling. Concurrently, disturbed shear facilitates nuclear translocation of the transcriptional co-activator YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif), further amplifying oxidative stress and endothelial dysfunction[74-76].
Recent mechanistic studies have delineated a Piezo1-soluble adenylyl cyclase (sAC)-Inositol 1,4,5-trisphosphate receptor 2 (IP3R2) axis, through which Piezo1 activation triggers endoplasmic reticulum (ER) Ca2+ release and Akt (also known as protein kinase B or PKB) signaling. Dysregulation of this pathway compromises eNOS phosphorylation and attenuates flow-mediated vasodilation, a hallmark of vascular aging[75]. In parallel, disturbed-flow-induced Piezo1/Ca2+ signaling engages Calmodulin (CaM)/Calcium/calmodulin-stimulated protein kinase II (CaMKII) and focal adhesion kinase pathways, enhancing YAP/TAZ nuclear translocation and reinforcing pro-inflammatory endothelial reprogramming[74,76].
Aging aggravates endothelial vulnerability through progressive glycocalyx thinning and shedding, heightened oxidative stress, cellular senescence, and altered vascular compliance. Together, these changes attenuate laminar shear-mediated signaling and shift mechanotransduction toward pro-atherogenic pathways[70,77]. These insights are informing translational strategies: pharmacological modulators of Piezo1 [e.g., Yoda1 (a Piezo1 agonist), Dooku1 (an analog of Yoda1, is a selective antagonist of the endogenous Piezo1 channel), GsMTx4 (Grammostola spatulata mechanotoxin 4, is a spider venom peptide that inhibits cationic mechanosensitive channels (MSCs), for example, TRPC1, TRPC6 and Piezo channels)] can modify endothelial Ca2+ entry and downstream signaling in experimental systems[78-80]. Targeting the Piezo1-sAC-IP3R2-Akt axis restores Akt/eNOS activation and promotes endothelial alignment in preclinical models, highlighting IP3R2-Akt stabilization as a potential therapeutic approach[75]. Concurrently, interventions aimed at preserving or restoring KLF2/KLF4 expression - via statins, metabolic modulators, or inhibition of negative regulators such as endothelial γ-protocadherins - show promise in reinstating NO-dependent vasoprotection in aging vasculature[73].
Collectively, these findings identify mechanosensitive channels (Piezo1), ER-Ca2+ regulatory pathways (IP3R2/sAC), and mechano-responsive transcriptional regulators (KLF2/KLF4, YAP/TAZ) as critical determinants of panvascular aging, delineating novel therapeutic targets for promoting protective NO-enriched endothelial phenotypes [Figure 2].
Inflammation/inflammaging
Aging-associated sterile inflammation, termed “inflammaging”, results from multiple convergent mechanisms. These include immunosenescence, SASP, mitochondrial DAMPs that activate the inflammasome and Cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) signaling, and age-related gut microbiota alterations. Collectively, these factors disrupt endothelial homeostasis across all vascular beds - macrovascular, microvascular, and organ-specific - thereby promoting plaque formation, progression, and instability[81-85].
In arteries and arterioles, SASP-derived cytokines (e.g., IL-1β, IL-6, TNF), proteases, and pro-thrombotic mediators enhance leukocyte recruitment, impair eNOS signaling, and increase vascular stiffness. Within capillary networks - such as those in the coronary, cerebral, renal, and retinal circulations - these mediators compromise endothelial barrier function and accelerate microvascular rarefaction, which are central features of panvascular aging[81,82].
Mitochondrial DAMPs, including mitochondrial DNA (mtDNA), sustain innate immune activation via Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3 (NLRP3) inflammasome and cGAS-STING pathways, directly linking bioenergetic dysfunction with chronic endothelial injury and atherogenesis[83]. Parallelly, age-related gut dysbiosis elevates pro-atherogenic metabolites (e.g., trimethylamine N-oxide, TMAO) and endotoxemia while reducing protective short-chain fatty acids (SCFAs), exacerbating endothelial dysfunction and arterial stiffness. Emerging human cohort data indicate that a “younger” gut microbial age correlates with lower cardiovascular risk in older adults, highlighting a modifiable gut-vascular aging axis[84,85].
A significant advance in recent years is the recognition that clonal hematopoiesis of indeterminate potential (CHIP) acts as a systemic, panvascular inflammatory driver. CHIP - particularly mutations in Tet methylcytosine dioxygenase 2 (TET2) and DNA Methyltransferase 3 Alpha (DNMT3A) - reprograms myeloid cells toward a hyperinflammatory phenotype characterized by increased cytokine production, thereby accelerating atherosclerosis and thrombosis. Multi-ancestry cohorts studies and mechanistic models have established CHIP as an age-related cardiovascular risk factor[86-88]. Notably, low-dose colchicine can partially attenuate CHIP-driven inflammation and atherogenesis in preclinical TET2-deficient models, aligning with its established clinical benefits in atherosclerotic disease[89].
Therapeutically, the CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) trial (NCT01327846) provided proof-of-concept that targeting IL-1β reduces recurrent cardiovascular events, with subsequent analyses confirming decreased total cardiovascular events and clarifying residual inflammatory risk mediated by upstream and downstream cytokines such as IL-18 and IL-6[90]. Low-dose colchicine has since emerged as a clinically actionable anti-inflammatory therapy. The Low-Dose Colchicine 2 (LoDoCo2) trial (2020; Australian New Zealand Clinical Trials Registry; ACTRN12614000093684) demonstrated a reduction in major adverse cardiovascular events (MACE) in patients with stable coronary disease. Together with the Colchicine Cardiovascular Outcomes Trial (COLCOT; NCT02551094), these findings supported the 2023 approval by the U.S. Food and Drug Administration (FDA) of low-dose colchicine (0.5 mg; Lodoco®) to reduce the risk of myocardial infarction, stroke, coronary revascularization, and cardiovascular death in eligible adults[91,92]. Beyond reducing clinical events, these interventions target shared inflammatory pathways that drive panvascular aging - offering a mechanistic strategy applicable across diverse vascular territories [Figure 2].
Oxidative stress
Excessive ROS production and mitochondrial dysfunction form a self-reinforcing cycle, further aggravated by eNOS uncoupling. Together, these processes induce endothelial damage, accelerate cellular senescence, and promote widespread vascular remodeling.
Aging-related defects in mitochondrial dynamics - marked by reduced fusion [mediated by Mitofusin 1 and 2 (MFN1/2) and Optic Atrophy 1 (OPA1)], enhanced fission [(via Dynamin-related protein 1 (DRP1)], and impaired mitophagy - lead to the accumulation of damaged mitochondria. This elevates mitochondrial ROS, activates redox-sensitive signaling pathways such as NF-κB, and induces SASP, all of which contribute to vascular stiffening and structural remodeling[93,94]. Concurrently, eNOS uncoupling diminishes NO bioavailability while generating additional ROS, further exacerbating endothelial dysfunction and senescence[95].
Therapeutic strategies targeting these interconnected pathways are increasingly being explored. Mitochondria-targeted antioxidants such as Mitoquinone (MitoQ) directly neutralize mitochondrial ROS and have been shown in preclinical studies to improve endothelial function, reduce arterial stiffness, and delay cellular senescence; early clinical trials also suggest beneficial vascular effects[96,97]. Similarly, interventions that restore NAD+ levels - through supplementation with precursors such as nicotinamide mononucleotide (NMN) or nicotinamide riboside (NR) - reactivate sirtuin signaling, enhance autophagy and mitochondrial biogenesis, suppress inflammation, and improve endothelial and VSMCs function in both animal models and early human studies[98,99].
In summary, the mutually reinforcing interplay among ROS overproduction, mitochondrial dysfunction, and eNOS uncoupling plays a central role in panvascular aging, driving endothelial injury, senescence, and maladaptive vascular remodeling. Therapeutic strategies focused on restoring mitochondrial homeostasis and NAD+ levels therefore represent promising approaches to attenuate vascular aging and its systemic consequences [Figure 2].
VSMCS DYSFUNCTION
Aging drives a panvascular remodeling process in which inflammatory and oxidative stress signals shift VSMCs from a contractile phenotype toward synthetic, osteogenic/chondrogenic, foam cell-like, fibroblast-like, and macrophage-like phenotypes[100,101]. This phenotypic switch promotes VSMCs proliferation, migration, ECM deposition, and secretion of cytokines and MMPs, collectively contributing to arterial stiffening and medial degeneration. Single-cell and lineage-tracing studies in aging models, including naturally aged mice, progeroid models, and chronically stressed arterial constructs, have shown that these transitions are driven by DNA-damage responses, mitochondrial ROS, and SASP cytokines. These factors activate KLF4/TCF21 programs, WNT (WNT signaling pathway)-RUNX2 (RUNX Family Transcription Factor 2, is a direct target of the canonical WNT signaling pathway) osteogenic signaling, and stress-kinase pathways [e.g., Receptor-Interacting Serine/Threonine-Protein Kinase 1 (RIPK1)] while suppressing contractile gene networks[100,102].
Across vascular beds, the cumulative outcome is loss of medial elasticity, fibrotic matrix accumulation, and calcification, resulting in conduit artery stiffening and impaired microvascular compliance - hallmarks of panvascular aging that propagate end-organ injury in the heart, brain, kidney, and retina[103-107]. Mechanotransduction reinforces these changes: increased ECM stiffness and disturbed hemodynamics activate YAP/TAZ and Transient receptor potential channels (TRP) channel signaling, further promoting synthetic/osteogenic reprogramming[103,104]. Conversely, epigenetic regulators such as TET2 and N6-methyladenosine (m6A) readers [e.g., YTH N6-Methyladenosine RNA Binding Protein F2 (YTHDF2)] can restrain osteogenic drift[105,106]. Clinically, these alterations manifest as elevated pulse pressure, isolated systolic hypertension, vascular calcification, aneurysm susceptibility, and vulnerable atherosclerotic plaques throughout coronary, carotid, cerebral, renal, and peripheral arteries, representing an integrated “panvascular” phenotype of aging[107-109].
Notably, emerging evidence indicates that pre-senescent VSMCs - transitioning toward senescence and exhibiting heightened DNA damage susceptibility - contribute to plaque destabilization and amplify neointimal hyperplasia after vascular injury, even before full expression of canonical senescence
Senotherapeutic approaches are advancing from concept to mechanism: in preclinical studies, dasatinib plus quercetin (D+Q) modifies chromatin organization in senescent VSMCs, reduces vascular senescence markers, and improves vasomotor function. However, its effects on plaque burden and stability remain context-dependent, varying with dosage and timing of administration[112-114]. Other emerging strategies aim to restore VSMCs homeostasis and attenuate the stiffness-calcification axis in panvascular aging.
TELOMERE SHORTENING & CELLULAR SENESCENCE
Cellular senescence of ECs, VSMCs, pericytes, and adventitial fibroblasts serves as a systemic driver of panvascular aging[117]. These senescent cells accumulate throughout the macro- and microvasculature and secrete a pro-inflammatory, matrix-remodeling SASP, characterized by elevated levels of IL-6, IL-1β, MMPs, chemokines, and growth factors. Through paracrine and endocrine signaling, the SASP disseminates dysfunction across the vascular wall and to peripheral organs[118]. This cascade leads to endothelial barrier dysfunction, impaired angiogenesis, maladaptive VSMC phenotypic switching (e.g., toward synthetic/osteogenic), ECM degradation and stiffening, vascular calcification, and systemic complications such as neurovascular and renal degeneration[41]. Recent studies further indicate that endothelial and pericyte senescence disrupt neurovascular coupling and BBB integrity[119], while senescent VSMCs and fibroblasts exacerbate arterial fibrosis and calcification via SASP-MMP circuits and epigenetic reprogramming[120]. Together, this establishes cellular senescence as a central mechanistic link in panvascular pathogenesis.
Both telomere attrition and epigenetic remodeling contribute to aging in a tissue-specific manner, with evidence pointing to amplified effects within the vasculature. First, ECs and VSMCs face unique extrinsic stresses - including pulsatile hemodynamic shear, transmural pressure, and a highly oxidative/inflammatory local milieu. These changes accelerate telomere dysfunction and DNA damage responses compared to many quiescent parenchymal cells. Endothelial telomere uncapping, for example, triggers a senescent, pro-inflammatory and pro-thrombotic phenotype that directly impairs vasodilation and barrier integrity[121,122]. Second, vascular cells exhibit marked phenotypic plasticity: VSMCs can undergo clonal expansion and phenotype switching in disease contexts, processes tightly regulated by locus-specific epigenetic changes (e.g., DNA methylation, histone modifications and chromatin accessibility) that amplify pathologic remodeling, such as neointima formation and calcification[108,123]. Third, epigenetic signatures and methylation-based “clocks” reveal vascular-enriched alterations that correlate with clinical outcomes such as arterial stiffness and atherosclerosis, suggesting that vascular epigenetic remodeling is not merely a bystander of systemic aging but an active contributor to vascular pathology[124,125]. Collectively, these findings indicate that telomere shortening, and epigenetic remodeling exert disproportionately profound functional consequences in vascular tissues due to (i) unique extrinsic exposures, (ii) cell-type-specific responses (senescence/SASP and VSMC plasticity), and (iii) locus-specific epigenetic reprogramming that promotes maladaptive remodeling.
These insights have spurred the development of “senotherapeutic” strategies. Senolytics agents which selectively eliminate senescent cells - most notably the combination of dasatinib and quercetin (D+Q) and the flavonoid fisetin - have advanced from preclinical vascular models to early-phase human trials[126]. In idiopathic pulmonary fibrosis, an open-label pilot study suggested that D+Q can improve physical function, prompting further controlled feasibility studies[127]. In early Alzheimer’s disease, a phase I open-label trial of D+Q demonstrated central nervous system penetration with mixed biomarker signals but acceptable short-term safety[128]. Additional feasibility studies in broader geriatric and disease-specific populations are ongoing[129].
Concurrently, senomorphic approaches - which aim to attenuate the SASP without inducing cell death - are being explored for vascular applications. These include JAK/STAT (Janus kinase/signal transducer and activator of transcription signaling pathway) inhibition and interventions targeting metformin- or mTOR-related pathways. Evidence indicates that modulating IL-6-JAK signaling or alleviating mitochondrial and telomere stress can suppress senescence programs in ECs and VSMCs[130-133]. Together, these findings position senolytics and senomorphics as promising therapeutic avenues for lowering or partially reversing panvascular aging, while underscoring the need for longer-term, randomized controlled trials to establish efficacy, define relevant vascular endpoints, and evaluate long-term safety [Figure 2].
ECM REMODELING & ARTERIAL STIFFNESS
The progressive stiffening of arteries, a hallmark of panvascular aging, results from interrelated structural alterations in the vascular wall. Key contributors include the accumulation of AGEs, elastin fragmentation, increased collagen deposition, and an imbalance between MMPs and tissue inhibitor of metalloproteinases (TIMPs). Together, these changes disrupt the biomechanical properties of arteries, reducing elasticity and impairing vascular compliance across both large and small vessels.
AGE crosslinking not only directly stiffens ECM but also promotes oxidative stress and inflammatory signaling, thereby accelerating vascular senescence and fibrosis[134]. Concurrently, elastin degradation and collagen deposition form a self-reinforcing cycle that further increases arterial stiffness and exacerbates hemodynamic stress[107]. Dysregulation of the MMP-TIMP system also plays a critical role, driving maladaptive ECM turnover that contributes to plaque instability in large arteries and microvascular rarefaction in small vessels[135].
Clinically, arterial stiffening is increasingly recognized as a central mechanism linking vascular aging to end-organ damage. Stiffened conduit arteries transmit excessive pulsatile energy into distal microvascular beds, promoting BBB dysfunction, glomerular injury, and retinal microangiopathy. These processes help explain the association between arterial stiffness and age-related conditions such as hypertension, CKD, and cognitive decline[136-138]. At the cellular level, increased mechanical stress from a remodeled ECM activates mechanosensitive pathways - including YAP/TAZ and Piezo1 signaling in VSMCs and ECs. This reinforces maladaptive gene expression patterns that perpetuate vascular dysfunction and structural remodeling[139].
Current treatment strategies are shifting from purely symptomatic management toward directly targeting the structural drivers of vascular stiffness. In addition to established approaches such as AGEs breakers, elastin stabilizers, and MMP/TIMP modulators, emerging interventions include senotherapeutics, epigenetic modifiers, and lifestyle interventions such as regular exercise and dietary AGEs restriction. Together, these multimodal strategies highlight the potential not only to attenuate arterial stiffness but in some cases, partially reverse it - offering a promising avenue for mitigating systemic vascular aging and its associated organ dysfunction [Figure 2].
EPIGENETICS & NON-CODING RNAS
A growing body of research demonstrates that epigenetic mechanisms, including DNA methylation, histone methylation/acetylation, and emerging acyl modifications, coordinate endothelial and VSMC senescence throughout the vasculature, driving features of panvascular aging such as arterial stiffening, macrovascular and microvascular rarefaction, and atherogenesis. Aberrant DNA methylation and histone post-translational modifications (PTMs) reprogram chromatin at pro-inflammatory and pro-senescence loci in vascular cells. For example, recent studies link altered methylation and acetylation states to endothelial dysfunction and VSMCs senescence, which in turn promotes plaque progression[140,141].
Novel histone acyl marks such as lactylation and succinylation integrate cellular metabolism with chromatin regulation in vascular aging. Histone lactylation increases in response to elevated glycolytic flux and directly activates pro-senescence and pro-atherogenic transcriptional programs. In VSMCs, Tumor Necrosis Factor Receptor-Associated Protein 1 (TRAP1)-mediated metabolic reprogramming elevates lactate levels, leading to histone deacetylase 3-dependent histone H4 lysine 12 lactylation (H4K12la) that enriches SASP gene promoters and exacerbates VSMCs senescence and atherosclerosis[142]. In ECs, glycolysis-driven histone H3 lysine 18 lactylation (H3K18la), mediated by a P300 (E1A‐binding protein P300)/ASF1A (anti‐silencing function 1A) complex, upregulates SNAI1 (Snail family transcriptional repressor 1) expression and promotes endothelial-to-mesenchymal transition (EndMT), thereby accelerating atherogenesis[143]. Succinylation also plays a critical regulatory role: endothelial Sirtuin 5 (SIRT5), a desuccinylase, restrains apoptosis by removing succinylation marks [e.g., at ERO1A-K396 (Lysine 396 (K396) residue of the Endoplasmic Reticulum Oxidoreductase 1 Alpha (ERO1A) protein)], revealing a succinylation-stress axis that likely contributes to vascular aging pathways[144].
Beyond chromatin-level control, noncoding RNAs systemically modulate these processes. miR-34a is consistently implicated in endothelial and VSMCs senescence (often acting through SIRT1 and Notch signaling) and worsens vascular remodeling, a role supported by recent human and experimental studies[145,146]. miR-146a functions as a key negative regulator of NF-κB-driven vascular inflammation, with emerging evidence indicating that modulating its activity can blunt innate inflammatory signaling and suppress atherogenesis[147,148]. Conversely, the endothelial-enriched miR-126 promotes vascular repair and limits endothelial activation; translational studies suggest that restoring miR-126 activity helps stabilize plaques and improves endothelial homeostasis in atherosclerosis[149,150]. Together, these epigenetic and microRNA networks integrate metabolic stress, chromatin remodeling, and inflammatory signaling across diverse vascular beds, collectively advancing panvascular progression from endothelial dysfunction to plaque growth and arterial stiffening with age [Figure 2].
BRAIN-VASCULAR AXIS
Aging profoundly affects the neurovascular unit (NVU), the multicellular functional module composed of cerebral ECs, pericytes, astrocytes, and neurons, which collectively maintains BBB integrity and cerebral homeostasis. With advancing aging, the BBB undergoes a gradual functional decline, characterized by increased permeability resulting from endothelial and pericyte senescence, loss of tight and adherens junction proteins, basement membrane remodeling, and chronic neuroinflammation[8,151]. Senescent cerebral microvascular ECs and pericytes not only exhibit impaired proliferative and barrier-forming capacities but also secrete pro-inflammatory and matrix-remodeling SASP factors, further weakening vascular integrity[152].
This age-dependent BBB dysfunction leads to two main consequences: (1) impaired clearance of neurotoxic proteins such as amyloid-β and tau, and (2) increased transendothelial passage of plasma-derived proteins and peripheral immune mediators into the central nervous system. Together, these mechanisms form a pathophysiological link between systemic vascular aging - characterized by arterial stiffening, endothelial dysfunction, and chronic low-grade inflammation - and the development of neurodegenerative disorders, including Alzheimer’s disease and vascular dementia[153,154].
Recent advances in clinical neuroimaging have shown that subclinical, region-specific BBB breakdown - detectable via biomarkers such as soluble platelet-derived growth factor receptor-β (sPDGFRβ) or dynamic contrast-enhanced magnetic resonance imaging (MRI) - often occurs in cognitively normal older adults and can precede the onset of noticeable cognitive decline[155,156]. Thus, BBB dysfunction serves as both an early indicator and a potential therapeutic target within the broader framework of panvascular aging, highlighting the pivotal role of neurovascular senescence in mediating bidirectional communication between peripheral vascular injury and brain pathology [Figure 2].
PANVASCULAR AGING IN MAJOR DISEASES
Cardiovascular diseases
Panvascular aging describes the systemic decline of vascular integrity that affects the entire arterial system, from large elastic arteries to arterioles and capillary networks. At the macrostructural level, aging-driven remodeling- characterized by elastin fragmentation, collagen cross-linking, and medial calcification- leads to progressive arterial stiffening. This is clinically reflected in elevated pulse-wave velocity (PWV) and widened pulse pressure, both established independent predictors of cardiovascular morbidity and mortality[157,158].
At the microvascular level, reduced NO bioavailability, mitochondrial dysfunction, and impaired endothelial mechanosensing limit vasodilatory capacity and capillary recruitment, contributing to myocardial supply-demand imbalance and tissue ischemia[12,159]. Together, these processes underlie the pathogenesis of coronary artery disease, accelerate atherogenesis, and play a particularly detrimental role in heart failure with preserved ejection fraction (HFpEF), where microvascular inflammation, capillary rarefaction, and endothelial senescence are now recognized as central pathological features[160]. Increased arterial stiffness, a hallmark of aging, also critically impairs coronary perfusion. Normally, the compliant aorta buffers the pulse wave generated by ventricular ejection. In a stiffened aorta, however, the reflected wave returns prematurely, reducing diastolic coronary perfusion. This hemodynamic alteration, indicated by increased PWV, can lead to myocardial dysfunction and remodeling[161-163]. It is important to note that this phenomenon is not well replicated in standard mouse models.
Mechanistically, aging endothelium exposed to disturbed shear stress undergoes metabolic reprogramming, which disrupts the NAD+-sirtuin-mitochondrial axis and alters glycolysis-lipid signaling crosstalk. This metabolic shift promotes endothelial senescence and the secretion of a SASP, which is enriched in interleukins (e.g., IL-6), MMPs, and pro-thrombotic mediators[30,164]. These inflammatory and proteolytic factors destabilize atherosclerotic plaques, increasing the risk of rupture and thrombosis. Importantly, intrinsic vascular aging mechanisms act synergistically with traditional risk factors such as dyslipidemia, hypertension, and diabetes, creating a feed-forward cycle that accelerates arterial stiffening, plaque vulnerability, and cardiovascular events[107].
A growing body of clinical evidence demonstrates that biomarkers of panvascular aging - especially arterial stiffness indices, carotid structural measures, and circulating vascular-inflammatory protein signatures - robustly predict cardiovascular morbidity and mortality. Carotid-femoral PWV, the gold-standard measure of central arterial stiffness, consistently predicts MACE, independent of traditional risk factors[7,165]. Similarly, estimated PWV from large population cohorts improves risk stratification beyond the traditional models and correlates with subclinical coronary atherosclerosis and incident atrial fibrillation[166,167]. Carotid intima-media thickness (CIMT) and carotid plaque burden further reflect structural aging of the vasculature and predict ischemic stroke and coronary events in a dose-dependent manner[168]. Recent plasma proteomic studies add a systemic dimension, showing that age-associated vascular signatures, which are enriched for complement activation, matrix remodeling, and endothelial injury pathways, strongly correlate with incident cardiovascular disease and all-cause mortality[169,170]. Panvascular aging represents not only a localized decline in vascular resilience but also a systemic driver of cardiovascular disease progression, integrating macrovascular stiffening and microvascular dysfunction into a unified pathogenic continuum.
Neurodegenerative diseases
The NVU of the brain - composed of ECs, pericytes, astrocytes, and neurons - ages in concert with the systemic vasculature. Consequently, key features of panvascular aging, such as endothelial dysfunction, ECM stiffening, and chronic low-grade inflammation, are also evident at the BBB[12,171]. These alterations progressively impair BBB integrity and transport function, weaken neurovascular coupling, and diminish perfusion reserve. As a result, they hinder the clearance of amyloid-β and metabolic waste, elevate susceptibility to microhemorrhage, and promote neuroinflammation - mechanisms that critically link vascular aging to Alzheimer’s disease and vascular dementia[8,155]. Recent studies highlight that biophysical remodeling of the BBB, including basement membrane stiffening, glycocalyx degradation, and impaired shear sensing, reshapes endothelial phenotype and transporter regulation, thereby exacerbating barrier leakage and neurovascular uncoupling[151,172]. Importantly, pericyte dysfunction destabilizes endothelial tight junctions and increases vesicular transcytosis, further compromising barrier function in aged cerebral vasculature[173]. Mechanistically, these insights reveal potential therapeutic strategies, aimed at restoring shear-dependent endothelial signaling, stabilizing pericyte-endothelial interactions, and preserving glycocalyx integrity, which have shown promise in preclinical models[62,174]. Clinically, neuroimaging and longitudinal biomarker studies demonstrate that BBB leakage increases with age and correlates with cognitive impairment. This supports the concept that cognitive decline reflects, at least in part, a manifestation of panvascular failure within the cerebral microcirculation[175,176]. Moreover, some biomarkers of vascular aging are strongly linked to neurodegenerative outcomes, including cognitive decline, cerebral small-vessel disease, and dementia.
Arterial stiffness has emerged as a key systemic driver of impaired cerebral perfusion and white-matter injury. Higher PWV is associated with reduced cerebral blood flow, increased white matter hyperintensity burden, and accelerated cognitive decline in aging populations[177-179]. Carotid structural indices, such as CIMT, carotid plaque burden, and carotid distensibility, also predict incident dementia and Alzheimer’s disease, illustrating how extracranial vascular aging contributes to intracranial microvascular dysfunction[180,181]. Furthermore, plasma proteomic signatures of aging, enriched for pathways involving vascular inflammation, BBB dysfunction, and complement cascades, are strongly correlated with future cognitive impairment, brain atrophy, and neurodegenerative disease onset[13]. Collectively, these findings indicate that neurodegeneration often develops within a systemic vascular aging milieu, wherein deteriorating vascular integrity across organs - detectable through accessible biomarkers - can help forecast long-term neurological outcomes.
CKD
The kidney, as one of the most highly vascularized organs, is particularly vulnerable to systemic vascular aging. Progressive stiffening and stenosis of renal arteries and arterioles transmit excessive pulsatile energy to the glomerular microcirculation, leading to glomerulosclerosis, EC senescence, oxidative stress, and capillary rarefaction[182,183]. As a consequence, the effective filtration surface area is reduced, while advancing tubulointerstitial fibrosis further accelerates the decline in glomerular filtration rate (GFR)[184].
Accumulating clinical and epidemiological evidence supports a bidirectional kidney-vascular axis, in which systemic vascular stiffness exacerbates renal injury, while CKD in turn amplifies vascular aging. Central arterial stiffness, commonly assessed by PWV, is a robust predictor of incident CKD and accelerated GFR decline across diverse populations[185,186]. Increased pulsatile load transmitted to the renal microvasculature is associated with impaired autoregulation, progressive albuminuria, and glomerulosclerosis. Consistently, cohort studies demonstrate that indices of arterial stiffness - including cardio-ankle vascular index (CAVI) and carotid-femoral PWV - predict renal functional decline even in individuals without overt hypertension or diabetes[157,,187].
Beyond hemodynamic measures, structural markers of arterial aging, such as increased CIMT and reduced arterial distensibility, independently correlate with microalbuminuria and CKD progression[184]. In parallel, emerging plasma proteomic signatures of vascular aging - reflecting endothelial dysfunction, ECM remodeling, and inflammatory activation - are associated with future renal events and can predict rapid GFR decline years before clinical diagnosis[13,188].
Diabetes
Diabetes accelerates a panvascular aging phenotype affecting both micro- and macrocirculation. Sustained hyperglycemia and insulin resistance elevate endothelial oxidative stress, eNOS uncoupling, thereby reducing NO bioavailability[77,189]. Concurrently, the accumulation of AGEs stiffens the ECM and disrupts receptor-mediated signaling, worsening vascular rigidity and impairing cellular function[190,191]. Mechanistically, AGEs promote non-enzymatic cross-linking within collagen fibrils, increasing tissue brittleness and reducing deformability[77,189].
In parallel, enzymatic remodeling of the EG - marked by accelerated hyaluronan turnover, elevated heparanase activity, and syndecan cleavage - leads to thinning of this critical mechanotransductive layer. Loss of glycocalyx integrity impairs shear-stress sensing and NO production, further exacerbating endothelial dysfunction[192,193]. Hyperglycemia-driven oxidative stress also accelerates proteoglycan degradation, compounding glycocalyx loss in diabetes[193,194].
Together, these molecular and structural changes reflect a shared vascular aging pathway that is prematurely activated under diabetic metabolic stress. The resulting panvascular phenotype manifests as the full spectrum of diabetic angiopathies - including retinopathy, nephropathy, neuropathy, peripheral artery disease, and coronary artery disease - reflecting coordinated dysfunction across diverse vascular beds[189].
Therapeutically, strategies aimed at preserving glycocalyx integrity and reducing AGEs accumulation are emerging as panvascular protective approaches. Nutritional and pharmacological interventions targeting glycocalyx regeneration - such as supplementation with glycocalyx precursors (e.g., glucosamine sulfate, high-molecular-weight hyaluronan) - have shown promise in preclinical diabetes models and are under clinical evaluation[195,196]. Experimental studies also indicate that co-treatment with agents such as albumin-bound S1P and heparan sulfate derivatives can synergistically enhance EG restoration[197]. In clinical settings, dietary pattern interventions, such as the Mediterranean diet, adequate vitamin D and omega-3 intake, and limitation of dietary AGEs, may also support glycocalyx health and vascular resilience[191].
In summary, diabetic vascular aging is driven by interconnected mechanisms involving oxidative stress, eNOS dysfunction, AGE-mediated matrix stiffening, and glycocalyx degradation. A comprehensive therapeutic strategy that combines glycocalyx-focused interventions, AGEs reduction, and conventional glycemic and blood-pressure control may offer a unified approach to attenuating panvascular aging in diabetes[192,197].
TRANSLATIONAL STAGING OF CANDIDATE ANTI-PANVASCULAR-AGING STRATEGIES
Translational readiness varies considerably among different approaches targeting panvascular aging. Broadly, these strategies can be categorized into five groups: (1) pharmacologic removal or modulation of senescent cells (senolytics and senomorphics), (2) metabolic restoration (particularly NAD+ boosting), (3) glycocalyx restoration, (4) non-pharmacologic interventions (lifestyle modification, structured risk-factor management, and device- or rehabilitation-based therapies), and (5) epigenetic modulators (particularly Epigenetic Clocks).
Senolytic and senomorphic agents
These compounds have demonstrated robust efficacy in animal models of vascular dysfunction. For example, the combination of D+Q reduces senescent cell burden, improves endothelium-dependent vasodilation, and attenuates arterial stiffening in aged mice. Senomorphic agents such as rapamycin and metformin suppress the SASP and ameliorate microvascular rarefaction[198,199]. Emerging human data from early-phase studies indicate that short-course senolytic therapy can reduce senescence markers in conditions such as idiopathic pulmonary fibrosis and diabetic kidney disease[126,200]. Small pilot trials of D+Q and other agents have shown feasibility and preliminary safety, though definitive efficacy data remain limited. Thus, senolytics currently occupy an early clinical translational stage (phase I-II exploratory studies)[132,201].
NAD+-boosting strategies
Approaches that elevate NAD+ levels - including supplementation with NR, NMN, and related compounds - are supported by extensive preclinical evidence for improving cellular energetics and vascular function. Several randomized and crossover human trials have demonstrated increases in circulating NAD+ metabolites and promising physiologic signals, positioning NAD+ boosters at a late-preclinical to early-clinical stage. The next necessary step is adequately powered phase II trials with clinical endpoints. Caution is warranted, however, as formulations, dosing, and downstream effects vary across studies[202,203].
Glycocalyx restoration strategies
Interventions aimed at restoring the EG - such as sulodexide, heparan-mimetic compounds, or agents that enhance GAG synthesis - have shown significant improvements in endothelial barrier integrity, capillary perfusion, and inflammatory signaling in preclinical models[204,205]. Clinically, sulodexide has been shown to improve the sublingual perfused boundary region (a surrogate for glycocalyx thickness) and enhance microvascular perfusion in patients with metabolic syndrome or early atherosclerosis[206].
Lifestyle and non-pharmacologic interventions
Established lifestyle measures - including aerobic and resistance exercise, dietary modification, smoking cessation, and blood pressure and lipid control, already occupy an implementation and population-health stage, given their proven ability to reduce cardiovascular events. However, their specific capacity to reverse molecular hallmarks of panvascular aging remains an active translational question, best addressed by incorporating mechanistic biomarker outcomes within pragmatic clinical trials[207].
Epigenetic clocks as biomarkers for anti-panvascular trials
Epigenetic clocks, which are defined by algorithms that estimate biological age from DNA methylation patterns - offer a quantifiable biomarker for monitoring intervention efficacy. They integrate cumulative molecular changes correlated with morbidity and mortality and can be measured longitudinally in accessible tissues such as blood. DNA methylation age acceleration is associated with vascular pathology and predicts cardiovascular outcomes, supporting their biological plausibility as surrogate endpoints in vascular-focused trials[208].
However, several considerations are important before adopting epigenetic clocks as primary trial biomarkers: First, many clocks are sensitive to shifts in cellular composition; methodological advances that correct for cell type or employ tissue-specific clocks are needed[209]. Second, clock performance and clinical relevance vary by clock type and population; pre-specification and validation in the target cohort are essential. Third, investigators should combine multiple complementary aging measures (e.g., epigenetic clocks plus vascular imaging, endothelial function, and established biomarkers), use longitudinal sampling with standardized assays, consider tissue- or pathway-informed clocks to increase sensitivity to vascular interventions, and power studies to detect meaningful changes that correlate with clinical outcomes[210].
In summary, epigenetic clocks are promising monitoring tools but should initially be deployed as exploratory endpoints within well-controlled trials that pair methylation-based metrics with functional vascular outcomes and rigorous assay standardization. Only through such iterative validation can they meet the evidentiary threshold required for regulatory or clinical decision-making[209,210].
Recommendations for trial design
For investigators planning anti-panvascular aging trials, based on our current knowledge, we recommend a multi-modular approach including: beginning with mechanism-focused phase I/II studies that combine (a) targeted biomarkers (e.g., senescence markers, NAD+ metabolites), (b) epigenetic clock panels as exploratory quantitative biomarkers, and (c) vascular physiological measures (e.g., PWV, FMD). This can be followed by measuring signals in later, larger, event-driven phase III programs.
CONCLUSION AND FUTURE PERSPECTIVES
Panvascular aging constitutes a systemic failure of the vascular hierarchy - spanning conduit, resistance, and capillary vessels - rather than a mere byproduct of chronological aging. Common pathogenic drivers, including endothelial dysfunction, mitochondrial impairment, NAD+ depletion, oxidative stress, low-grade inflammation, ECM remodeling, and cellular senescence, converge across diverse clinical spectrums such as cardiovascular disease, neurodegeneration, and CKD. This shared vascular foundation suggests that local injuries are not isolated; rather, epigenetic and metabolic dysregulation allow vascular decline to propagate multisystem effects, ultimately driving multimorbidity.
Consequently, the therapeutic paradigm must shift from organ- specific interventions toward integrated, system-level approaches. Promising strategies involve restoring metabolic homeostasis - - specifically via NAD+ boosters and senolytics - alongside lifestyle modifications to ensure sustained protection. However, translating these insights into clinical practice requires addressing critical knowledge gaps.
Future research must prioritize the development of cross-organ biomarker panels - integrating proteomics and epigenetic clocks - to quantify systemic biological age. Furthermore, longitudinal cohorts utilizing harmonized imaging (e.g., cfPWV, retinal Optical coherence tomography angiography (OCTA), and hemodynamic MRI) are essential to identify signatures that predict multi-organ decline. By leveraging large-animal models and organ-on-chip technologies, the field can better dissect inter-organ communication. Ultimately, establishing a coherent framework for panvascular biology will depend on standardized composite endpoints and biomarker-stratified clinical trials to reverse coordinated vascular decline across the lifespan.
DECLARATIONS
Acknowledgments
All figures were created using https://www.biorender.com/, and publication rights were obtained accordingly.
Authors’ contributions
Made substantial contributions to the conception, writing, and editing of the manuscript: Yalan W, Yu TX
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Hong Kong Research Grants Council General Research Fund (14107822, 14105321); Young Collaborative Research Grant (C4004-23Y); Theme-based Research Scheme (T12-101/23-N); CUHK Group Research Scheme; CUHK Lo Kwee-Seong Biomedical Research Fund (Startup); the National Natural Science Foundation of China (32071145); the Science and Technology Planning Project of Guangdong Province, China (2023B1212120009); the Natural Science Foundation of Hunan Province (2024JJ6490); the Fundamental Research Funds for the Central Universities (5022044016); and the Changsha Municipal Natural Science Foundation (kq2402232).
Conflicts of interest
Tian XY is an Editorial Board Member of the journal Vessel Plus, but was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. 2012;110:1097-108.
2. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. 2018;123:849-67.
4. Tanriover C, Copur S, Mutlu A, et al. Early aging and premature vascular aging in chronic kidney disease. Clin Kidney J. 2023;16:1751-65.
5. Fountoulakis N, Miyamoto Y, Pavkov ME, Karalliedde J, Maltese G. Pathophysiology of vascular ageing and the effect of novel cardio-renal protective medications in preventing progression of chronic kidney disease in people living with diabetes. Diabet Med. 2025;42:e15464.
6. Mitchell GF, Hwang SJ, Vasan RS, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121:505-11.
7. Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55:1318-27.
8. Montagne A, Barnes SR, Sweeney MD, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296-302.
9. Sabbatinelli J, Prattichizzo F, Olivieri F, Procopio AD, Rippo MR, Giuliani A. Where metabolism meets senescence: focus on endothelial cells. Front Physiol. 2019;10:1523.
10. Pacinella G, Ciaccio AM, Tuttolomondo A. Endothelial dysfunction and chronic inflammation: the cornerstones of vascular alterations in age-related diseases. Int J Mol Sci. 2022;23:15722.
11. Lacolley P, Regnault V, Segers P, Laurent S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol Rev. 2017;97:1555-617.
12. Ungvari Z, Tarantini S, Kiss T, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. 2018;15:555-65.
13. Oh HS, Rutledge J, Nachun D, et al. Organ aging signatures in the plasma proteome track health and disease. Nature. 2023;624:164-72.
14. Pilling LC, Atkins JL, Bowman K, et al. Human longevity is influenced by many genetic variants: evidence from 75,000 UK Biobank participants. Aging. 2016;8:547-60.
15. Grosse L, Wagner N, Emelyanov A, et al. Defined p16(High) senescent cell types are indispensable for mouse healthspan. Cell Metab. 2020;32:87-99.e6.
16. Kalucka J, de Rooij LPMH, Goveia J, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. 2020;180:764-779.e20.
17. Gulej R, Nyúl-Tóth Á, Csik B, et al. Rejuvenation of cerebromicrovascular function in aged mice through heterochronic parabiosis: insights into neurovascular coupling and the impact of young blood factors. Geroscience. 2024;46:327-47.
18. Loffredo FS, Steinhauser ML, Jay SM, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828-39.
19. Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90-4.
20. Chen X, Luo Y, Zhu Q, et al. Small extracellular vesicles from young plasma reverse age-related functional declines by improving mitochondrial energy metabolism. Nat Aging. 2024;4:814-38.
21. Yeboah J, Crouse JR, Hsu FC, Burke GL, Herrington DM. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults: the cardiovascular health study. Circulation. 2007;115:2390-7.
22. Tian YE, Cropley V, Maier AB, Lautenschlager NT, Breakspear M, Zalesky A. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat Med. 2023;29:1221-31.
23. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. 2018;123:825-48.
24. Wang M, Kim SH, Monticone RE, Lakatta EG. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension. 2015;65:698-703.
25. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018;14:576-90.
26. Mayer O, Gelžinský J, Seidlerová J, et al. The role of advanced glycation end products in vascular aging: which parameter is the most suitable as a biomarker? J Hum Hypertens. 2021;35:240-9.
27. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104-16.
28. Mencke R, Hillebrands JL. ; NIGRAM consortium. The role of the anti-ageing protein Klotho in vascular physiology and pathophysiology. Ageing Res Rev. 2017;35:124-46.
29. Toth L, Czigler A, Hegedus E, et al. Age-related decline in circulating IGF-1 associates with impaired neurovascular coupling responses in older adults. Geroscience. 2022;44:2771-83.
30. Childs BG, Li H, van Deursen JM. Senescent cells: a therapeutic target for cardiovascular disease. J Clin Invest. 2018;128:1217-28.
31. Ghebre YT, Yakubov E, Wong WT, et al. Vascular aging: implications for cardiovascular disease and therapy. Transl Med. 2016;6:183.
32. Jones RC, Karkanias J, Krasnow MA, et al. ; Tabula Sapiens Consortium*. The tabula sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376:eabl4896.
33. Rivera CF, Farra YM, Silvestro M, et al. Mapping the unicellular transcriptome of the ascending thoracic aorta to changes in mechanosensing and mechanoadaptation during aging. Aging Cell. 2024;23:e14197.
34. Li Y, Ren P, Dawson A, et al. Single-cell transcriptome analysis reveals dynamic cell populations and differential gene expression patterns in control and aneurysmal human aortic tissue. Circulation. 2020;142:1374-88.
35. Cheng J, Wu H, Xie C, et al. Single-cell mapping of large and small arteries during hypertensive aging. J Gerontol A Biol Sci Med Sci. 2024;79:glad188.
36. Kim SY, Cheon J. Senescence-associated microvascular endothelial dysfunction: a focus on the blood-brain and blood-retinal barriers. Ageing Res Rev. 2024;100:102446.
37. Fu X, Zhao Y, Cui X, et al. Cxcl9 modulates aging associated microvascular metabolic and angiogenic dysfunctions in subcutaneous adipose tissue. Angiogenesis. 2025;28:17.
38. Dobner S, Tóth F, de Rooij LPMH. A high-resolution view of the heterogeneous aging endothelium. Angiogenesis. 2024;27:129-45.
39. Liu Z, Huang Y, Wang D, et al. Insights gained from single-cell RNA analysis of murine endothelial cells in aging hearts. Heliyon. 2023;9:e18324.
40. Gao P, Gao P, Choi M, et al. Transcriptome analysis of mouse aortae reveals multiple novel pathways regulated by aging. Aging. 2020;12:15603-23.
41. Iwao T, Takata F, Matsumoto J, et al. Senescence in brain pericytes attenuates blood-brain barrier function in vitro: A comparison of serially passaged and isolated pericytes from aged rat brains. Biochem Biophys Res Commun. 2023;645:154-63.
42. Berthiaume AA, Schmid F, Stamenkovic S, et al. Pericyte remodeling is deficient in the aged brain and contributes to impaired capillary flow and structure. Nat Commun. 2022;13:5912.
43. Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12+ perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18:1262-70.
44. Dias Moura Prazeres PH, Sena IFG, Borges IDT, et al. Pericytes are heterogeneous in their origin within the same tissue. Dev Biol. 2017;427:6-11.
45. Yamazaki T, Nalbandian A, Uchida Y, et al. Tissue myeloid progenitors differentiate into pericytes through TGF-β signaling in developing skin vasculature. Cell Rep. 2017;18:2991-3004.
46. Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557-61.
47. Greenberg JI, Shields DJ, Barillas SG, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456:809-13.
48. Jiang L, Xu L, Mao J, et al. Rheb/mTORC1 signaling promotes kidney fibroblast activation and fibrosis. J Am Soc Nephrol. 2013;24:1114-26.
49. Bennett HC, Zhang Q, Wu YT, et al. Aging drives cerebrovascular network remodeling and functional changes in the mouse brain. Nat Commun. 2024;15:6398.
50. Shaito A, Aramouni K, Assaf R, et al. Oxidative stress-induced endothelial dysfunction in cardiovascular diseases. Front Biosci. 2022;27:105.
51. Hernandez-Navarro I, Botana L, Diez-Mata J, et al. Replicative endothelial cell senescence may lead to endothelial dysfunction by increasing the BH2/BH4 ratio induced by oxidative stress, reducing BH4 availability, and decreasing the expression of eNOS. Int J Mol Sci. 2024;25:9890.
52. Bouly M, Bourguignon MP, Roesch S, et al. Aging increases circulating BH2 without modifying BH4 levels and impairs peripheral vascular function in healthy adults. Transl Res. 2021;238:36-48.
53. Chuaiphichai S, Rashbrook VS, Hale AB, et al. Endothelial cell tetrahydrobiopterin modulates sensitivity to ang (angiotensin) ii-induced vascular remodeling, blood pressure, and abdominal aortic aneurysm. Hypertension. 2018;72:128-38.
54. Bisconti AV, Garten RS, Broxterman RM, et al. No effect of acute tetrahydrobiopterin (BH4) supplementation on vascular dysfunction in the old. J Appl Physiol. 2022;132:773-84.
55. Lin H, Pan J, Zhang J, et al. Intermedin protects peritubular capillaries by inhibiting eNOS uncoupling through AMPK/GTPCH-I/BH4 pathway and alleviate CKD following AKI. Free Radical Biol Med. 2025;234:72-85.
56. Du J, Zhu X, Zhang Y, et al. CTRP13 attenuates atherosclerosis by inhibiting endothelial cell ferroptosis via activating GCH1. Int Immunopharmacol. 2024;143:113617.
57. Rodríguez C, Muñoz M, Contreras C, Prieto D. AMPK, metabolism, and vascular function. FEBS J. 2021;288:3746-71.
58. Chang TT, Lin LY, Chen C, Chen JW. CCL4 contributes to aging related angiogenic insufficiency through activating oxidative stress and endothelial inflammation. Angiogenesis. 2024;27:475-99.
59. Yang Y, Chen H, Liu Q, Niu Y, Mao C, Wang R. Elucidating molecular pathogenesis and developing targeted therapeutic interventions for cerebrovascular endothelial cell-mediated vascular dementia. Front Aging Neurosci. 2025;17:1623050.
60. Li J, Wang Y, Shrestha S, Gewirtz AT, Ding Y, Zou J. Targeting Gut Microbiota to Combat Vascular Aging and Cardiovascular Disease: Mechanisms and Therapeutic Potential. Nutrients. 2025;17:2887.
61. Madeddu P, Goulopoulou S, Wambeke D. Integrating endothelial-derived hyperpolarizing signaling into a multitarget therapeutic strategy for microvascular disease. arXiv 2025;arXiv:2508.01618. Available from https://arxiv.org/abs/2508.01618 [accessed 2 March 2026].
62. Shi SM, Suh RJ, Shon DJ, et al. Glycocalyx dysregulation impairs blood-brain barrier in ageing and disease. Nature. 2025;639:985-94.
63. Milusev A, Rieben R, Sorvillo N. The Endothelial glycocalyx: a possible therapeutic target in cardiovascular disorders. Front Cardiovasc Med. 2022;9:897087.
64. Ying J, Zhang C, Wang Y, et al. Sulodexide improves vascular permeability via glycocalyx remodelling in endothelial cells during sepsis. Front Immunol. 2023;14:1172892.
65. Charfeddine S, Ibnhadjamor H, Jdidi J, et al. Sulodexide significantly improves endothelial dysfunction and alleviates chest pain and palpitations in patients with long-COVID-19: insights from TUN-EndCOV study. Front Cardiovasc Med. 2022;9:866113.
66. Pollmann S, Scharnetzki D, Manikowski D, Lenders M, Brand E. Endothelial dysfunction in Fabry disease is related to glycocalyx degradation. Front Immunol. 2021;12:789142.
67. Mishra S, Ipe U, Nottebaum AF, Peters KG, Vestweber D. Inhibition of VE-PTP rejuvenates Schlemm’s canal in aged mice and acts via Tie2. PLoS ONE. 2025;20:e0323615.
68. Zeng Y, Adamson RH, Curry FR, Tarbell JM. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am J Physiol Heart Circ Physiol. 2014;306:H363-72.
69. Souilhol C, Serbanovic-Canic J, Fragiadaki M, et al. Endothelial responses to shear stress in atherosclerosis: a novel role for developmental genes. Nat Rev Cardiol. 2020;17:52-63.
70. Wang X, Shen Y, Shang M, Liu X, Munn LL. Endothelial mechanobiology in atherosclerosis. Cardiovasc Res. 2023;119:1656-75.
71. Zheng Q, Zou Y, Teng P, et al. Mechanosensitive channel PIEZO1 senses shear force to induce KLF2/4 expression via CaMKII/MEKK3/ERK5 axis in endothelial cells. Cells. 2022;11:2191.
72. Dabravolski SA, Sukhorukov VN, Kalmykov VA, Grechko AV, Shakhpazyan NK, Orekhov AN. The role of KLF2 in the regulation of atherosclerosis development and potential use of KLF2-targeted therapy. Biomedicines. 2022;10:254.
73. Joshi D, Coon BG, Chakraborty R, et al. Endothelial γ-protocadherins inhibit KLF2 and KLF4 to promote atherosclerosis. Nat Cardiovasc Res. 2024;3:1035-48.
74. Barettino A, González-Gómez C, Gonzalo P, et al. Endothelial YAP/TAZ activation promotes atherosclerosis in a mouse model of Hutchinson-Gilford progeria syndrome. J Clin Invest. 2024;134:e173448.
75. Santana Nunez D, Malik AB, Lee Q, et al. Piezo1 induces endothelial responses to shear stress via soluble adenylyl cyclase-IP3R2 circuit. iScience. 2023;26:106661.
76. Yang Y, Wang D, Zhang C, et al. Piezo1 mediates endothelial atherogenic inflammatory responses via regulation of YAP/TAZ activation. Hum Cell. 2022;35:51-62.
77. Sun L, Wang L, Ye KX, et al. Endothelial glycocalyx in aging and age-related diseases. Aging Dis. 2023;14:1606-17.
78. Syeda R, Xu J, Dubin AE, et al. Chemical activation of the mechanotransduction channel Piezo1. Elife. 2015;4:e07369.
79. Wijerathne TD, Ozkan AD, Lacroix JJ. Yoda1’s energetic footprint on Piezo1 channels and its modulation by voltage and temperature. Proc Natl Acad Sci U S A. 2022;119:e2202269119.
80. Matrongolo MJ, Ang PS, Wu J, et al. Piezo1 agonist restores meningeal lymphatic vessels, drainage, and brain-CSF perfusion in craniosynostosis and aged mice. J Clin Invest. 2023;134:e171468.
81. Müller L, Di Benedetto S. Inflammaging, immunosenescence, and cardiovascular aging: insights into long COVID implications. Front Cardiovasc Med. 2024;11:1384996.
82. Kumar M, Yan P, Kuchel GA, Xu M. Cellular senescence as a targetable risk factor for cardiovascular diseases: therapeutic implications: JACC Family Series. JACC Basic Transl Sci. 2024;9:522-34.
83. Ueda K, Sakai C, Ishida T, et al. Cigarette smoke induces mitochondrial DNA damage and activates cGAS-STING pathway: application to a biomarker for atherosclerosis. Clin Sci. 2023;137:163-80.
84. Brunt VE, Gioscia-Ryan RA, Casso AG, et al. Trimethylamine-N-oxide promotes age-related vascular oxidative stress and endothelial dysfunction in mice and healthy humans. Hypertension. 2020;76:101-12.
85. Longtine AG, Greenberg NT, Bernaldo de Quirós Y, Brunt VE. The gut microbiome as a modulator of arterial function and age-related arterial dysfunction. Am J Physiol Heart Circ Physiol. 2024;326:H986-H1005.
86. Díez-Díez M, Ramos-Neble BL, de la Barrera J, et al. Unidirectional association of clonal hematopoiesis with atherosclerosis development. Nat Med. 2024;30:2857-66.
87. Oren O, Small AM, Libby P. Clonal hematopoiesis and atherosclerosis. J Clin Invest. 2024;134:e180066.
88. Rauch PJ, Gopakumar J, Silver AJ, et al. Loss-of-function mutations in Dnmt3a and Tet2lead to accelerated atherosclerosis and concordant macrophage phenotypes. Nat Cardiovasc Res. 2023;2:805-18.
89. Zuriaga MA, Yu Z, Matesanz N, et al. Colchicine prevents accelerated atherosclerosis in TET2-mutant clonal haematopoiesis. Eur Heart J. 2024;45:4601-15.
90. Everett BM, MacFadyen JG, Thuren T, Libby P, Glynn RJ, Ridker PM. Inhibition of interleukin-1β and reduction in atherothrombotic cardiovascular events in the CANTOS Trial. J Am Coll Cardiol. 2020;76:1660-70.
91. von Scheidt M, Adkar SS, Krefting J, et al. Clonal haematopoiesis of indeterminate potential and mortality in coronary artery disease. Eur Heart J. 2026;47:453-69.
92. FDA approval letter for Lodoco (colchicine) 0.5 mg (June 16, 2023); ACC perspective on the evolving role of colchicine (2025) Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/215727Orig1s000ltr.pdf [Last accessed on 12 Mar 2026].
93. Zhang Z, Zhao X, Zhang X, et al. Mitochondrial cardiovascular diseases: molecular mechanisms, multi-omics exploration and therapeutic strategies. J Adv Res. 2025.
94. Lin JR, Shen WL, Yan C, Gao PJ. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler Thromb Vasc Biol. 2015;35:1413-22.
95. Janaszak-Jasiecka A, Płoska A, Wierońska JM, Dobrucki LW, Kalinowski L. Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cell Mol Biol Lett. 2023;28:21.
96. Rossman MJ, Santos-Parker JR, Steward CAC, et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension. 2018;71:1056-63.
97. Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014;592:2549-61.
98. Yoshino M, Yoshino J, Kayser BD, et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science. 2021;372:1224-9.
99. Fang EF, Hou Y, Lautrup S, et al. NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat Commun. 2019;10:5284.
100. Chen R, McVey DG, Shen D, Huang X, Ye S. Phenotypic switching of vascular smooth muscle cells in atherosclerosis. J Am Heart Assoc. 2023;12:e031121.
101. Gong X, Liu Y, Liu H, et al. Re-analysis of single-cell transcriptomics reveals a critical role of macrophage-like smooth muscle cells in advanced atherosclerotic plaque. Theranostics. 2024;14:1450-63.
102. Li Y, Jie W, Qi Y, et al. Inhibition of RIPK1 alleviating vascular smooth muscle cells osteogenic transdifferentiation via Runx2. iScience. 2024;27:108766.
103. Jafarinia H, Khalilimeybodi A, Barrasa-Fano J, Fraley SI, Rangamani P, Carlier A. Insights gained from computational modeling of YAP/TAZ signaling for cellular mechanotransduction. NPJ Syst Biol Appl. 2024;10:90.
104. Ritsvall O, Albinsson S. Emerging role of YAP/TAZ in vascular mechanotransduction and disease. Microcirculation. 2024;31:e12838.
105. He D, Ma J, Zhou Z, et al. TET2 suppresses vascular calcification by forming an inhibitory complex with HDAC1/2 and SNIP1 independent of demethylation. J Clin Invest. 2025;135:e186673.
106. Li L, Cheng M, Jin J, et al. The m6A reader YTHDF2 protects vascular smooth muscle cells against the osteogenic differentiation through targeting Runx2. Renal Fail. 2025;47:2488876.
107. Herzog MJ, Müller P, Lechner K, et al. Arterial stiffness and vascular aging: mechanisms, prevention, and therapy. Signal Transduct Target Ther. 2025;10:282.
108. Lambert J, Jørgensen HF. Epigenetic regulation of vascular smooth muscle cell phenotypes in atherosclerosis. Atherosclerosis. 2025;401:119085.
109. Zhu C, Bishop T, Gregorich ZR, Guo W. Titin is a new factor regulating arterial stiffness through vascular smooth muscle cell tone in male rats. Physiol Rep. 2025;13:e70270.
110. Kaistha A, Oc S, Garrido AM, et al. Premature cell senescence promotes vascular smooth muscle cell phenotypic modulation and resistance to re-differentiation. Cardiovasc Res. 2025;121:1448-63.
111. Azar P, Jarr KU, Gomez D, Jørgensen HF, Leeper NJ, Bochaton-Piallat ML. Smooth muscle cells in atherosclerosis: essential but overlooked translational perspectives. Eur Heart J. 2025;46:4862-75.
112. Gadecka A, Nowak N, Bulanda E, et al. The senolytic cocktail, dasatinib and quercetin, impacts the chromatin structure of both young and senescent vascular smooth muscle cells. Geroscience. 2025;47:3907-25.
113. Rad AN, Grillari J. Current senolytics: mode of action, efficacy and limitations, and their future. Mech Ageing Dev. 2024;217:111888.
114. Zha Y, Zhuang W, Yang Y, Zhou Y, Li H, Liang J. Senescence in vascular smooth muscle cells and atherosclerosis. Front Cardiovasc Med. 2022;9:910580.
115. Cheng M, Jin J, Zhang D, et al. METTL3 obstructs vascular smooth muscle cells osteogenic reprogramming by methylating Runx2 in chronic kidney disease. Commun Biol. 2025;8:582.
116. Chen A, Yuan P, Lu Y, et al. Smooth muscle-specific HuR knockout attenuates vascular calcification. J Mol Cell Cardiol. 2025;205:117-28.
117. Bloom SI, Islam MT, Lesniewski LA, Donato AJ. Mechanisms and consequences of endothelial cell senescence. Nat Rev Cardiol. 2023;20:38-51.
118. Culley MK, Chan SY. Endothelial senescence: a new age in pulmonary hypertension. Circ Res. 2022;130:928-41.
119. van der Linden J, Stefens SJM, Heredia-Genestar JM, et al. Ercc1 DNA repair deficiency results in vascular aging characterized by VSMC phenotype switching, ECM remodeling, and an increased stress response. Aging Cell. 2024;23:e14126.
120. Stefens SJM, van Vliet N, IJpma A, et al. Increased vascular smooth muscle cell senescence in aneurysmal Fibulin-4 mutant mice. NPJ Aging. 2024;10:31.
121. Bloom SI, Liu Y, Tucker JR, et al. Endothelial cell telomere dysfunction induces senescence and results in vascular and metabolic impairments. Aging Cell. 2023;22:e13875.
122. Gao Z, Santos RB, Rupert J, et al. Endothelial-specific telomerase inactivation causes telomere-independent cell senescence and multi-organ dysfunction characteristic of aging. Aging Cell. 2024;23:e14138.
123. Chen Y, Liang L, Wu C, et al. Epigenetic control of vascular smooth muscle cell function in atherosclerosis: a role for DNA methylation. DNA Cell Biol. 2022;41:824-37.
124. Wang K, Liu H, Hu Q, et al. Epigenetic regulation of aging: implications for interventions of aging and diseases. Signal Transduct Target Ther. 2022;7:374.
125. Han Y, Kim SY. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp Mol Med. 2023;55:1-12.
126. Justice JN, Nambiar AM, Tchkonia T, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554-63.
127. Nambiar A, Kellogg D 3rd, Justice J, et al. Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. EBioMedicine. 2023;90:104481.
128. Gonzales MM, Garbarino VR, Kautz TF, et al. Senolytic therapy in mild Alzheimer’s disease: a phase 1 feasibility trial. Nat Med. 2023;29:2481-8.
129. Zhu X, Zhang C, Liu L, Xu L, Yao L. Senolytic combination of dasatinib and quercetin protects against diabetic kidney disease by activating autophagy to alleviate podocyte dedifferentiation via the Notch pathway. Int J Mol Med. 2024;53:26.
130. Atlante S, Gottardi Zamperla M, Cis L, Farsetti A, Gaetano C. Senolytic therapies for cardiovascular aging: tackling fibrosis and metabolic dysfunction. Eur J Intern Med. 2025;140:106413.
131. Yang B, Li T, Wang Z, et al. Ruxolitinib-based senomorphic therapy mitigates cardiomyocyte senescence in septic cardiomyopathy by inhibiting the JAK2/STAT3 signaling pathway. Int J Biol Sci. 2024;20:4314-40.
132. Lelarge V, Capelle R, Oger F, Mathieu T, Le Calvé B. Senolytics: from pharmacological inhibitors to immunotherapies, a promising future for patients’ treatment. NPJ Aging. 2024;10:12.
133. Tai S, Sun J, Zhou Y, et al. Metformin suppresses vascular smooth muscle cell senescence by promoting autophagic flux. J Adv Res. 2022;41:205-18.
134. Wang B, Jiang T, Qi Y, et al. AGE-RAGE axis and cardiovascular diseases: pathophysiologic mechanisms and prospects for clinical applications. Cardiovasc Drugs Ther. 2025;39:1489-506.
135. Castro MM, Rizzi E, Prado CM, Rossi MA, Tanus-Santos JE, Gerlach RF. Imbalance between matrix metalloproteinases and tissue inhibitor of metalloproteinases in hypertensive vascular remodeling. Matrix Biol. 2010;29:194-201.
136. Krishnan S, Suarez-Martinez AD, Bagher P, et al. Microvascular dysfunction and kidney disease: challenges and opportunities? Microcirculation. 2021;28:e12661.
137. Hanssen H, Streese L, Vilser W. Retinal vessel diameters and function in cardiovascular risk and disease. Prog Retin Eye Res. 2022;91:101095.
138. Rensma SP, Stehouwer CDA, Van Boxtel MPJ, et al. Associations of arterial stiffness with cognitive performance, and the role of microvascular dysfunction: the maastricht study. Hypertension. 2020;75:1607-14.
139. Sladitschek-Martens HL, Guarnieri A, Brumana G, et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature. 2022;607:790-8.
140. Zhang L, Xia C, Yang Y, et al. DNA methylation and histone post-translational modifications in atherosclerosis and a novel perspective for epigenetic therapy. Cell Commun Signal. 2023;21:344.
141. Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res. 2021;128:474-91.
142. Li X, Chen M, Chen X, et al. TRAP1 drives smooth muscle cell senescence and promotes atherosclerosis via HDAC3-primed histone H4 lysine 12 lactylation. Eur Heart J. 2024;45:4219-35.
143. Dong M, Zhang Y, Chen M, et al. ASF1A-dependent P300-mediated histone H3 lysine 18 lactylation promotes atherosclerosis by regulating EndMT. Acta Pharm Sin B. 2024;14:3027-48.
144. Pan Y, Zhang Q, Li C, et al. SIRT5 alleviates apoptosis of vascular endothelial cells under simulated microgravity via desuccinylation of ERO1A. Int J Mol Sci. 2025;26:2908.
145. Hua CC, Liu XM, Liang LR, Wang LF, Zhong JC. Targeting the microRNA-34a as a novel therapeutic strategy for cardiovascular diseases. Front Cardiovasc Med. 2021;8:784044.
146. Qian Z, Huang Y, Yang N, et al. miR-34a-5p/MARCHF8/ADAM10 axis in the regulation of vascular endothelial cell dysfunction and senescence. Mech Ageing Dev. 2025;225:112060.
147. Ardinal AP, Wiyono AV, Estiko RI. Unveiling the therapeutic potential of miR-146a: targeting innate inflammation in atherosclerosis. J Cell Mol Med. 2024;28:e70121.
148. Zhai K, Deng L, Wu Y, et al. Extracellular vesicle-derived miR-146a as a novel crosstalk mechanism for high-fat induced atherosclerosis by targeting SMAD4. J Adv Res. 2025;73:729-41.
149. Guo B, Gu J, Zhuang T, et al. MicroRNA-126: from biology to therapeutics. Biomed Pharmacother. 2025;185:117953.
150. Ye ZQ, Meng XH, Fang X, Liu HY, Mwindadi HH. MiR-126 regulates the effect of mesenchymal stem cell vascular repair on carotid atherosclerosis through MAPK/ERK signaling pathway. World J Stem Cells. 2025;17:106520.
151. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99:21-78.
152. Graves SI, Baker DJ. Implicating endothelial cell senescence to dysfunction in the ageing and diseased brain. Basic Clin Pharmacol Toxicol. 2020;127:102-10.
153. Gorelick PB, Scuteri A, Black SE, et al. ; American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42:2672-713.
154. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133-50.
155. Nation DA, Sweeney MD, Montagne A, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270-6.
156. Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J Exp Med. 2017;214:3151-69.
158. Ma S, Xie X, Yuan R, et al. Vascular aging and atherosclerosis: a perspective on aging. Aging Dis. 2024;16:33-48.
159. Kirkman DL, Robinson AT, Rossman MJ, Seals DR, Edwards DG. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2021;320:H2080-100.
160. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263-71.
161. Chirinos JA, Segers P, Hughes T, Townsend R. Large-artery stiffness in health and disease: JACC state-of-the-art review. J Am Coll Cardiol. 2019;74:1237-63.
162. Chirinos JA, Segers P. Noninvasive evaluation of left ventricular afterload: part 2: arterial pressure-flow and pressure-volume relations in humans. Hypertension. 2010;56:563-70.
163. Coutinho T, Mielniczuk LM, Srivaratharajah K, deKemp R, Wells GA, Beanlands RS. Coronary artery microvascular dysfunction: role of sex and arterial load. Int J Cardiol. 2018;270:42-7.
164. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. 2021;18:58-68.
165. Laurent S, Cockcroft J, Van Bortel L, et al. ; European Network for Non-invasive Investigation of Large Arteries. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588-605.
166. Vishram-Nielsen JKK, Laurent S, Nilsson PM, et al. ; MORGAM Project. Does estimated pulse wave velocity add prognostic information? MORGAM prospective cohort project. Hypertension. 2020;75:1420-8.
167. Ejiri K, Ding N, Kim E, et al. Association of segment-specific pulse wave velocity with vascular calcification: the ARIC (atherosclerosis risk in communities) study. J Am Heart Assoc. 2024;13:e031778.
168. O'Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson SK Jr. Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med. 1999;340:14-22.
169. Tanaka T, Basisty N, Fantoni G, et al. Plasma proteomic biomarker signature of age predicts health and life span. Elife. 2020;9:e61073.
170. Shah AM, Myhre PL, Arthur V, et al. Large scale plasma proteomics identifies novel proteins and protein networks associated with heart failure development. Nat Commun. 2024;15:528.
171. Soto-Rojas LO, Pacheco-Herrero M, Martínez-Gómez PA, et al. The neurovascular unit dysfunction in Alzheimer’s disease. Int J Mol Sci. 2021;22:2022.
172. Vanlandewijck M, He L, Mäe MA, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475-80.
173. Nikolakopoulou AM, Montagne A, Kisler K, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22:1089-98.
174. Yamamoto K, Nogimori Y, Imamura H, Ando J. Shear stress activates mitochondrial oxidative phosphorylation by reducing plasma membrane cholesterol in vascular endothelial cells. Proc Natl Acad Sci U S A. 2020;117:33660-7.
175. Nehra G, Bauer B, Hartz AMS. Blood-brain barrier leakage in Alzheimer’s disease: from discovery to clinical relevance. Pharmacol Ther. 2022;234:108119.
176. Montagne A, Barnes SR, Nation DA, Kisler K, Toga AW, Zlokovic BV. Imaging subtle leaks in the blood-brain barrier in the aging human brain: potential pitfalls, challenges, and possible solutions. Geroscience. 2022;44:1339-51.
177. Jefferson AL, Cambronero FE, Liu D, et al. Higher aortic stiffness is related to lower cerebral blood flow and preserved cerebrovascular reactivity in older adults. Circulation. 2018;138:1951-62.
178. Tomoto T, Tarumi T, Zhang R. Central arterial stiffness, brain white matter hyperintensity and total brain volume across the adult lifespan. J Hypertens. 2023;41:819-29.
179. Aimagambetova B, Ariko T, Merritt S, Rundek T. Arterial stiffness measured by pulse wave velocity correlated with cognitive decline in hypertensive individuals: a systematic review. BMC Neurol. 2024;24:393.
180. Wang W, Norby FL, George KM, et al. Association of carotid intima-media thickness and other carotid ultrasound features with incident dementia in the ARIC-NCS. J Am Heart Assoc. 2021;10:e020489.
181. Geijselaers SL, Sep SJ, Schram MT, et al. Carotid stiffness is associated with impairment of cognitive performance in individuals with and without type 2 diabetes. The Maastricht study. Atherosclerosis. 2016;253:186-93.
182. O'Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005;46:200-4.
183. Sedaghat S, Mattace-Raso FU, Hoorn EJ, et al. Arterial stiffness and decline in kidney function. Clin J Am Soc Nephrol. 2015;10:2190-7.
184. Daehn IS, Duffield JS. The glomerular filtration barrier: a structural target for novel kidney therapies. Nat Rev Drug Discov. 2021;20:770-88.
185. Yao Z, Ishigami J, Kim E, et al. Arterial stiffness and subsequent incidence of CKD and Kidney function decline in a large longitudinal community cohort: the atherosclerosis in communities (ARIC) study. Am J Kidney Dis. 2025;86:32-42.
186. Liu JJ, Liu S, Lee J, et al. Aortic pulse wave velocity, central pulse pressure, augmentation index and chronic kidney disease progression in individuals with type 2 diabetes: a 3- year prospective study. BMC Nephrol. 2020;21:359.
187. Nagayama D, Fujishiro K, Miyoshi T, et al. Predictive ability of arterial stiffness parameters for renal function decline: a retrospective cohort study comparing cardio-ankle vascular index, pulse wave velocity and cardio-ankle vascular index 0. J Hypertens. 2022;40:1294-302.
188. Grams ME, Surapaneni A, Chen J, et al. Proteins associated with risk of kidney function decline in the general population. J Am Soc Nephrol. 2021;32:2291-302.
189. Li Y, Liu Y, Liu S, et al. Diabetic vascular diseases: molecular mechanisms and therapeutic strategies. Signal Transduct Target Ther. 2023;8:152.
190. Chaudhuri J, Bains Y, Guha S, et al. The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality. Cell Metab. 2018;28:337-52.
191. Franceković P, Gliemann L. Endothelial glycocalyx preservation-impact of nutrition and lifestyle. Nutrients. 2023;15:2573.
192. Banerjee S, Mwangi JG, Stanley TK, Mitra R, Ebong EE. Regeneration and assessment of the endothelial glycocalyx to address cardiovascular disease. Ind Eng Chem Res. 2021;60:17328-47.
193. Wang J, Ma L, Fang Y, Ye T, Li H, Lan P. Factors influencing glycocalyx degradation: a narrative review. Front Immunol. 2024;15:1490395.
194. Gimblet CJ, Ernst JW, Bell B, et al. Effect of glycocalyx-targeted therapy on vascular function in older adults: a randomized controlled trial. J Appl Physiol. 2024;136:1488-95.
195. Smith JA, Ramirez-Perez FI, Burr K, et al. Impact of dietary supplementation of glycocalyx precursors on vascular function in type 2 diabetes. J Appl Physiol. 2024;137:1592-603.
196. ClinicalTrials.gov. Targeting the endothelial glycocalyx to enhance vascular function and exercise-induced vascular adaptations in type 2 diabetes. Available from: https://clinicaltrials.gov/study/NCT05205005 [Last accessed on 12 Mar 2026].
197. Mitra R, Pentland K, Kolev S, et al. Co-therapy with S1P and heparan sulfate derivatives to restore endothelial glycocalyx and combat pro-atherosclerotic endothelial dysfunction. Life Sci. 2025;377:123662.
198. Islam MT, Hall SA, Dutson T, et al. Endothelial cell-specific reduction in mTOR ameliorates age-related arterial and metabolic dysfunction. Aging Cell. 2024;23:e14040.
199. Moiseeva O, Deschênes-Simard X, St-Germain E, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell. 2013;12:489-98.
200. Hickson LJ, Langhi Prata LGP, Bobart SA, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446-56.
201. Suda M, Paul KH, Tripathi U, Minamino T, Tchkonia T, Kirkland JL. Targeting Cell senescence and senolytics: novel interventions for age-related endocrine dysfunction. Endocr Rev. 2024;45:655-75.
202. Freeberg KA, Udovich CC, Martens CR, Seals DR, Craighead DH. Dietary supplementation with NAD+-boosting compounds in humans: current knowledge and future directions. J Gerontol A Biol Sci Med Sci. 2023;78:2435-48.
203. Henderson JD, Quigley SNZ, Chachra SS, Conlon N, Ford D. The use of a systems approach to increase NAD+ in human participants. NPJ Aging. 2024;10:7.
204. Broekhuizen LN, Lemkes BA, Mooij HL, et al. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia. 2010;53:2646-55.
205. Gil N, Goldberg R, Neuman T, et al. Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes. 2012;61:208-16.
206. Ikonomidis I, Pavlidis G, Lambadiari V, et al. Endothelial glycocalyx and microvascular perfusion are associated with carotid intima-media thickness and impaired myocardial deformation in psoriatic disease. J Hum Hypertens. 2022;36:1113-20.
207. González LDM, Romero-Orjuela SP, Rabeya FJ, Del Castillo V, Echeverri D. Age and vascular aging: an unexplored frontier. Front Cardiovasc Med. 2023;10:1278795.
208. Sánchez-Cabo F, Fuster V, Silla-Castro JC, et al. Subclinical atherosclerosis and accelerated epigenetic age mediated by inflammation: a multi-omics study. Eur Heart J. 2023;44:2698-709.
209. Tomusiak A, Floro A, Tiwari R, et al. Development of an epigenetic clock resistant to changes in immune cell composition. Commun Biol. 2024;7:934.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].