
Overcoming tumor antigen heterogeneity in CAR-T cell therapy for malignant mesothelioma (MM)

 , ...
, ... Abstract
Malignant mesothelioma (MM) is a rare, aggressive solid tumor with limited therapeutic options and poor therapeutic response. The role of immunotherapy in MM is now well established and therapeutic options, such as checkpoint inhibitors, are increasingly being approved. Chimeric antigen receptor (CAR)-T cell therapy is successfully implemented in several hematologic cancers, but currently has inadequate effect in solid tumors, owing to several limitations, such as trafficking and infiltration, limited T cell persistence and exhaustion, the immunosuppressive TME and tumor antigen heterogeneity. The lack of uniform and universal expression of tumor-associated antigens (TAAs) on tumor cells, as well as TAA heterogeneity following tumor editing post-therapy, are issues of significant importance to CAR-T cell and associated antigen-targeting therapies. Our review discusses the concept of tumor antigen heterogeneity in MM, the consequences for CAR-T cell therapies and the strategies to overcome it.
Keywords
INTRODUCTIONS
Malignant mesothelioma (MM) is a rare, highly aggressive malignancy of the mesothelial cells lining the body’s serosal membranes. The currently well-known cause for the development of MM is exposure to asbestos, with radiation and BAP-1 germline mutations also being implicated as possible causes[1]. Most patients (~75%) are diagnosed with malignant pleural mesothelioma (MPM), which affects the mesothelial lining of the lung. Around 20% of mesothelioma diagnoses are peritoneal mesothelioma (MPeM) developing in the peritoneal membrane. Less than 5% of all MM cases primarily develop in the pericardial membrane, or in the tunica vaginalis in men. Despite treatment advances in recent years, prognosis remains poor, with a median overall survival (OS) of approximately one year when treated with pemetrexed-platinum chemotherapy and a 5-year survival of around 10%[2,3].
The pemetrexed-platinum combination chemotherapy has remained the mainstay of treatment in MPM since its introduction 17 years ago and until 2021[4-6]. The addition of the anti-angiogenic agent bevacizumab to pemetrexed/cisplatin chemotherapy improved MPM OS in selected patients from 16.1 to 18.8 months[7,8], but has not been widely adopted. The multimodality treatment of surgery, radiotherapy, and pemetrexed/cisplatin chemotherapy has resulted in 5-year survival of only about 5% and is a viable option for only a small number of MPM patients[9,10]. MM’s advanced stage, localization, and morphology contribute to its high resistance to therapy. Therefore, there has been a need for more efficient therapies to be developed and tried in this disease. In recent years, several novel immunotherapies have been developed and tested in clinical trials, including chimeric antigen receptor (CAR)-T cell immunotherapies[11,12].
CAR-T cell therapy for MM involves directly targeting tumor surface antigens like the glycoprotein mesothelin or targeting the tumor stroma through antigens expressed by cancer-associated fibroblasts (CAFs). The development of efficacious CAR-T cell therapies for advanced solid tumors like MM presents several significant challenges relating to efficient CAR-T cell trafficking to and infiltration in the tumor, limited CAR-T cell persistence and exhaustion, and a highly immunosuppressive tumor microenvironment (TME). Another important challenge is tumor antigen heterogeneity, which refers to the varying and diverse levels of tumor-associated antigen (TAA) expression across different patients with the same cancer or even within the same patients at different time points[13,14].
In this review, we provide an in-depth overview of all aspects of tumor antigen heterogeneity and its implications in the development of CAR-T cell therapies against MM. Furthermore, we discuss the novel, innovative CAR-T cell approaches that could significantly contribute to overcoming tumor antigen heterogeneity, therefore offering CAR-T cells the opportunity to attack and eliminate MM and other solid tumors more efficiently.
CURRENT TREATMENT LANDSCAPE IN MM
Pemetrexed/cisplatin chemotherapy, the mainstay of MPM treatment, has been combined with targeted therapies, such as tyrosine kinase inhibitors (TKIs) erlotinib and gefitinib and the mTOR inhibitor everolimus, but have been so far unsuccessful[15-18]. The treatment for MPeM, according to the National Comprehensive Cancer Network (NCCN), is first-line platinum-pemetrexed chemotherapy; beyond the first-line, there has not been any FDA-approved or established treatment regimen for MPeM. MPeM treatment strategies vary based on the patients’ disease progression state and consensus regarding the optimal treatment regimen for MPeM is still lacking. More recently, single-agent immune checkpoint inhibitor (ICI) immunotherapy targeting programmed cell death ligand 1 (PD-L1) (NCT01772004) or programmed death protein 1 (PD-1) (NCT02399371, NCT02054806, NCT02991482 NCT02716272) have demonstrated a modest increase in the median progression-free survival (PFS) in MM ranging from 2.5 to 6.2 months, but a slight improvement in overall survival[19,20]. The combination of nivolumab (anti-PD-1 antibody) plus ipilimumab (anti-CTLA-4 antibody) (NCT03048474, NCT02716272, NCT02899299) resulted in a PFS of 4.8-6.8 months (OS 15.9-18.1) and further improved MPM response rates (RR) from 25.8 to 29% as compared with anti-PD1/ anti-PD-L1 monotherapy[20]. Of note, the MPM patients who seemed to benefit the most were those with sarcomatoid histology[21]. The promising results of opilimumab/nivolumab combination therapy led to the approval of this regime for use as first-line therapy for MPM. As second-line therapy for MPM, gemcitabine and/or vinorelbine or ICI therapies are used[22-25]. Figure 1 outlines the currently available therapeutic options for MPM, with the timeline representing the median OS.
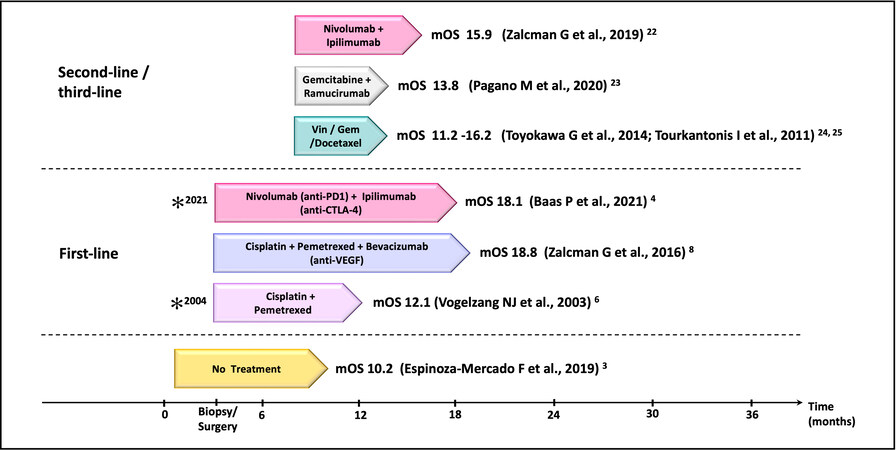
Figure 1. The current therapeutic landscape of malignant pleural mesothelioma (MPM). A list of the first and second/third-line therapies in relation to their median overall survival (mOS). *FDA/EMA/NICE approval (for Nivolumab-Ipilimumab, FDA approval was granted in 2020, and EMA/NICE approval in 2021). Vin: Vinorelbine; Gem: gemcitabine.
In a study by Kim et al., ICIs appeared to benefit patients with advanced MPM after initial platinum-based chemotherapy[26]. These findings, however, could not be extrapolated in the PROMISE-meso phase III randomized clinical trial, which failed to demonstrate the survival benefit of pembrolizumab over gemcitabine or vinorelbine[27]. Resistance to ICIs is multifactorial, involving the TME, the intrinsic tumoral signaling and host genetic factors of which the primary resistance to ICIs in MPM is attributed to the moderate to low levels of PD-L1 expression with low tumor mutational burden (TMB)[28]. Compared with MPM, MPeM has a slightly higher PD-L1 and TMB, and this difference in biology may unravel the ICI therapy[29]. Despite the clinical, molecular and epidemiological differences, the MPeM treatment strategies are largely based on the MPM studies, that is retrospective evidence-based. The overall low response rate for the second-line treatment drugs warrants further research and patients' referrals for participating in clinical trials.
CAR-T CELL THERAPY IN MM
Cellular immunotherapy, or adoptive cell therapy, are treatments that utilize the body’s immune cells to fight cancer. Several cellular immunotherapies exist, such as engineered T cell receptor (TCR) therapy, tumor-infiltrating lymphocyte (TIL) therapy, and chimeric antigen receptor T (CAR-T) cell therapy, all demonstrating feasibility of adoptive transfer and treatment successes, especially in hematological malignancies[30].
Cellular immunotherapy research in MM has been mainly focused on CAR-T cell therapy. CAR-T cells are patient autologous T cells that are genetically re-engineered ex-vivo to express a non-MHC-restricted receptor (CAR) targeting a specific tumor cell surface antigen. The CAR is composed of an antibody-derived single-chain variable fragment (scFv) fused to a flexible spacer/hinge region, a transmembrane part, and an intracellular signaling domain (CD3-ζ or Fc-γ)[31,32]. The CD3ζ intracellular signaling domain of CARs (or else, 1st generation CARs) was shown to provide limited therapeutic efficacy in initial trials[33], so CAR technology evolved to 2nd and 3rd generation CARs. These newer generation CARs incorporated additional co-stimulatory molecules, such as CD28, 4-1BB, or OX40, to the CD3ζ primary signaling domain and exhibited enhanced cell proliferation and greater antitumor potency due to increased signaling strength[34]. To date, all the available FDA/EMA/NICE-approved CAR-T cell therapies are either
CAR-T cell therapies against tumor-associated antigens (TAAs) or stroma-associated proteins have been developed and trialed in MM. Mesothelin (MSLN) is a cell-surface glycoprotein involved in tumor invasion and is expressed in 85% to 90% of MMs, with its expression elsewhere limited to the mesothelial surfaces at low levels[35-37]. Previous research involving MSLN-knockout mice established no histologic or anatomic abnormalities, suggesting MSLN may be non-essential for normal mouse development or reproduction[38], making it a great therapeutic target. The majority of current CAR-T cell trials target MSLN in MPM[39] , few in MPeM, and a range of solid tumors[40]. Other CAR-T cell targets explored in preclinical studies of MM include the ErbB family of receptors[41], the oncofetal glycoprotein 5T4[42], chondroitin sulfate proteoglycan 4 (CSPG4)[43], MET[44] and alkaline phosphatase placental-like 2 (ALPE2)[45].
In addition to directly targeting the tumor, CARs that target the tumor-associated stroma are gaining importance in CAR-T cell therapy, and are of great interest in fibrotic tumors like MM. Fibroblast activation protein (FAP) is a stromal antigen present on cancer-associated fibroblasts (CAFs), endothelial cells, and on the surface of some mesothelioma cells[46,47]. FAP-expressing CAFs are present in all three major MPM subtypes, so a number of preclinical studies and a clinical trial investigated FAP CAR-T cells’ therapeutic potential in MM[48,49]. Table 1 summarises the ongoing CAR-T cell therapy trials in MM.
CAR-T cell clinical trials in malignant mesothelioma (MM)
| Sl. no. | Target(s) | Study description | Phase | Clinical trial number | Sponsor/collaborator |
| 1. | MSLN | anti-MSLN CAR-T cell therapy with anti-PD1 | I | NCT04577326r | • Memorial Sloan-Kettering Cancer Center • Atara Biotherapeutics |
| 2. | MSLN PD-1 | anti-MSLN CAR-T cells secreting PD-1 nanobodies | I | NCT04489862r | • Wuhan Union Hospital, China • Shanghai Cell Therapy GroupCo., Ltd |
| 3. | MSLN | anti-MSLN CAR-T cell (intraperitoneal MCY-M11 - non-viral, mRNA-based) | I | NCT03608618* | • MaxCyte, Inc. • CTI Clinical Trial and Consulting Services |
| 4. | MSLN | anti-MSLN CAR-T cells (single retroviral vector) | I/II | NCT01583686* | • National Cancer Institute (NCI) • National Institutes of Health Clinical Center (CC) |
| 5. | MSLN | anti- MSLN CAR-T cells (lentiviral transduced anti-MSLN immunoreceptor SS1 fused to the 4-1BB and CD3ζ signaling domains) | I | NCT02159716c | • University of Pennsylvania |
| 6. | FAP | anti-FAP CAR-T cells (retroviral vector) | I | NCT01722149c | • University of Zurich |
| 7. | MSLN | anti-MSLN CAR-T cells (local delivery, lentiviral transduced) | I | NCT03054298r | • University of Pennsylvania • National Institutes of Health (NIH) • Tmunity Therapeutics |
| 8. | MSLN | anti-MSLN CAR-T cells (iCasp9M28z CAR-transduced) | I/II | NCT02414269a,nr | • Memorial Sloan-Kettering Cancer Center • Bellicum Pharmaceuticals • United States Department of Defence |
| 9. | MSLN | anti-MSLN CAR-T cells | I | NCT02580747un | • Chinese PLA General Hospital |
| 10. | MSLN | anti-MSLN CAR-T cells (mRNA-based, 4-1BB and CD3ζ signaling domains) | I | NCT01355965c | • University of Pennsylvania |
| 11. | MSLN, CD19, CD22, CD33, CD38, NY-ESO-1, DR5, C-Met, EGFR V III, | anti-MSLN, anti-CD19; anti-CD22; anti-CD33 ; anti-CD38; anti-NY-ESO-1; anti-DR5; anti-C-met; anti-EGFR V III; (multi-target gene-modified immunotherapy, CAR-T/TCR-T cells include ten different tumour-specific antibodies) | I/II | NCT03638206r | • Shenzhen BinDeBio Ltd. • The First Affiliated Hospital of Zhengzhou University |
There are several challenges that CAR-T cell therapy faces in solid tumors, like mesothelioma. These include (i) poor CAR-T cell trafficking to the tumor site and reduced infiltration in the tumor; (ii) the immunosuppressive effects of the tumor microenvironment (TME); (iii) upregulation of inhibitory receptors; (iv) CAR-T cell reduced proliferative ability, limited persistence and exhaustion. In addition, tumor antigen heterogeneity plays a significant role in efficacious CAR-T cell therapy.
TUMOR ANTIGEN HETEROGENEITY
The expression of tumor cell surface antigens, like CD19, in hematological malignancies is relatively uniform and stable. In contrast, solid tumor cell surface antigens are rarely universally expressed; instead, they present heterogeneity in their distribution and concentration, and are termed tumor-associated antigens (TAAs). TAA heterogeneity is a significant yet understudied issue in CAR-T- cell therapy for solid tumors[13].
Tumor heterogeneity in MM has been observed at many levels that overall contribute to the tumor’s aggressiveness and pose an obstacle to therapy. Intertumor heterogeneity is common in MM, with molecular analyses revealing an epithelioid-sarcomatoid type between patients’ tumors rather than distinct subclasses, which correlates strongly with molecular markers of the epithelial-mesenchymal transition[50]. The interpatient heterogeneity of MM has hampered clinical trial design and results. Comprehensive genomic and transcriptomic analysis of MM has revealed that “one-size-fits-all” approaches to therapy are unsuccessful due to extensive genomic heterogeneity among MM patients. Inactivated tumor suppressor genes, including BAP1, NF2, CDKN2A, and SETD2, dominate the mutational landscape in MM[51]. Intratumoral heterogeneity, meaning the presence of distinct tumor cell populations within the same tumor, is also observed in MM. In a study using whole-exome sequencing in MPM samples of several patients and from three different locations (anterior, posterior and diaphragm positions), distinct mutational patterns at the three different locations with a high degree of intratumoral spatial heterogeneity by varying amounts of subclonal fractions were found[52]. In addition to this, the TCRβ sequencing and immune gene expression revealed that this heterogeneity also extended to the TME[52]. These intratumoral differences in the TME may result in treatment resistance[53]. Finally, the temporal MPM heterogeneity is another issue - tumors can evolve during multiple rounds of cell division, and the presence of selective treatment with an anti-cancer drug further drives the tumor clonal evolution[54]. This temporal heterogeneity has important implications for treatment decisions and treatment outcomes.
Immunoproteogenomic analysis of MPM suggested the requirement for high measured abundance of neopeptides in the presence of high expression of MHC proteins specific for these neopeptides that predicted response to ICI treatment in MPM patients[55]. There is also evidence that MPM patients post-dasatinib treatment presented with higher T-cell clonality and a higher portion of T cells/T cell response across all tumor regions, which possibly led to their significantly longer survival and a clear sign of the extremely heterogeneous TCR repertoire in MPM[56]. A study by Sneddon et al. supported the existence of candidate neoantigens in MM that induced specific T-cell response to a set of predicted neoantigens from MPM effusions despite the low mutation burden of tumor[57]; these neoantigens may provide an actionable target/vaccine candidates for personalized therapeutics in difficult to treat cancers like MM, and the selection of patients to receive immunotherapies. Whereas the findings of another study by Mansfield et al. represented the discovery of potential neoantigen expression driven by structural chromosomal rearrangements[58], these results may have implications for developing novel immunotherapeutic strategies and selecting patients to receive immunotherapies.
TAA HETEROGENEITY AND CARs
Contrasting with acute lymphoblastic leukaemia (ALL) and other hematologic malignancies, TAA heterogeneity in solid tumors hampers effective CAR-T cell therapy. The inherent heterogeneous expression of the TAAs, combined with immune escape caused by low antigen levels on tumor cells, is the leading cause of resistance to CAR-T-cells and determines the efficacy of CAR-T cell therapy[59,60]. Antigen heterogeneity is defined as the differential expression of a number of TAAs, resulting in variation in the tumor cell phenotype and distribution of tumor antigen-positive cells. MM has both tumor cell and TME heterogeneity[61], and clinical studies of TAAs have observed the outgrowth of variants that lose the target antigen due to significant heterogeneity within the tumors. Hence, screening and stratifying patients based on the percentage of target antigen-expressing cells is essential, and selecting the patients over certain threshold levels of antigen-expressing cells for therapy[62]. For CAR-T cell therapy, choosing a target with high specificity and stability in addition to the high density of antigen-expressing cells within the tumor would play a vital role in the tumor cell lysis[63].
Role of intrinsic activity of CARTs
The critical determinants of CAR-T cell therapy-induced antitumor responses are CAR-T expression density on T cells and the affinity of CAR-T cells to recognize TAAs at even low expression levels. The affinity of CAR-T cells is determined by their binding kinetics and the rates at which they associate with and dissociate from their targets. The T cell receptors (TCRs) usually have a higher affinity to foreign antigens than the TAAs, yet the TCRs can recognize very low levels of antigens via the serial triggering mechanism[64]. On the other hand, higher levels of TAA density are required by the CAR-T cells for their activation, commonly known as the activating threshold[65], which induces cytokine production and cell proliferation as opposed to triggering cytolytic activity (lytic threshold)[66,67]. The expression of CARs on the surface of T cells affects the CAR-T cell activation; lower levels of CAR expression results in sub-activation of the CAR-T cells, while the overexpression of CARs can induce antigen-independent activation, accelerating cell differentiation and exhaustion, or apoptosis[68]. Avidity or functional avidity generally refers to the interaction between the receptor and the antigen. Avidity is the multimeric interaction between the tumor cell and the T-cell, whereas functional avidity is the functional response of a T cell (e.g., cytokine secretion or cytotoxicity)[69]. An overall increase in the affinity or avidity of CAR-T cells typically induces a more robust response[70]. Furthermore, tumor antigen-mediated downregulation of CARs also affects CAR density on the cell surface[71]. This downregulation of CAR on the cell surface is independent of CAR affinity and associated with T cell CAR density and TAA density[72]. CAR scFvs, in general, have a higher affinity to the target antigen compared to the affinity of TCRs. Considering the majority of the TAAs are self-antigens that are overexpressed on tumor cells and are weakly expressed on normal cells, there is a tendency for the high-affinity scFv CARs to attack normal cells[73]. This on-target, off-tumor toxicity may raise safety concerns and is one of the inevitable and common adverse effects of CAR-T cell therapy. The new generation of the CARs has modified scFvs with optimized affinity, enabling CAR-T cells to favorably attack tumor cells with overexpressed TAAs[74]. Remarkably, it is now possible to generate a large pool of antibodies with a wide range of affinity, each targeting the same epitope with a different affinity performed by light chain exchange technology. This approach provides a feasible option to screen a number of scFvs simultaneously and enables determining the best scFv affinity required for the activation of the CAR-T cell[75]. Therefore, understanding the affinity and interaction between the TAA density on tumor cells and the scFv density on the CAR-T cells will contribute to developing strategies for better CAR-T cell therapies.
Tumor antigen escape
TAA heterogeneity and/or high mutation rates are linked to the tumor antigen escape from immune recognition, which occurs by modulation of targeted TAA expression. In solid tumors, loss of target antigen and/or mutations in the target antigen are frequent observations post CAR-T cell infusion[76]; in such cases, employing an additional target is one possible solution. Even in hematological cancers, patients have exhibited resistance to CAR-T cell therapy and the emergence of tumor antigen escape variants is a primary concern, accounting for about 7%-25% of relapsed patients post therapy with CD19-targeting CAR-T cells[59]. The most common mechanisms of this include antigen loss due to mutations, splice variants of the gene, and lineage switching from CD19+ to CD19- (lymphoid to myeloid malignancy)[77]. In the case of solid tumors with an inherent heterogeneous expression of the tumor antigens, antigen escape is further exacerbated[60]. One of the studies in this field found that heterogeneous expression of TAA EGFRvIII and the inhibitory TME, which becomes even more immunosuppressive after CAR-T cell infusion, were significant barriers to the clinical efficacy of CAR-T cell therapy[76]. While the heterogeneous expression of TAAs could be overcome by CARs with dual or triple antigen targeting, the TME would require drug targeting against its immunosuppressive components. In a recent study by Larson et al., a genome-wide CRISPR knockout screen was conducted in glioblastoma, in which CAR-T cell therapy has limited efficacy, and it was found that the loss of interferon-γ receptor (IFNγR) signaling pathway genes, such as JAK1 or JAK2 and IFNGR1, rendered glioblastoma and other solid tumors more resistant to killing by CAR-T cells both in vitro and in vivo[78]. This study further reported that the defect in IFNGR1 signaling is one of the important mechanisms of tumor cell-intrinsic resistance to CAR-T cells, which may require improved binding interactions between the CAR-T cells and the tumor cells for better outcomes in solid tumors[78].
APPROACHES TO OVERCOME TAA HETEROGENEITY
Several modifications to the CAR design and translational strategies have aided in overcoming TAA heterogeneity to some extent in recent years. First is the choice of the target antigen, which can be focused on the ones associated with tumor invasion or metastasis, so the CAR-T cells generated are directed to more aggressive cancer cells. Another strategy is targeting multiple antigens or having the CAR deliver “payloads” that then kill tumor cells in different ways, potentially by inducing a broader immune response in the patient. Novel CAR designs, such as multi-antigen targeted CARs (dual/triple TAA-targeted CARs), tandem CARs (TanCARs), armored CARS, switchable CARs, and synNotch CARs in combination with inhibitory CARs are currently being investigated and are discussed in this review[79-85].
Novel CAR strategies
Tumor cells expressing low levels of a targeted TAA may survive following CAR-T cell therapy, as opposed to those with high levels of the TAA that are preferentially eliminated[86,87]. Therefore, solid tumors that are heterogeneous in nature may not benefit from single-antigen targeting CAR-T cell therapy. Previous studies have confirmed the efficiency of CAR-T cell therapy that simultaneously targets two TAAs, for example, MSLN and EpCAM[88], or MSLN and MUC16[89]. To further widen the spectrum of antitumor function, CARs can be modified to produce inhibitory peptides, cytokines, chemokine receptors, and immunomodulatory agents to target the TME; these are known as armored CARs[90,91]. The inhibitory CAR (iCAR) fuses an antigen recognition domain, mostly an antigen expressed on normal tissue, with an inhibitory intracellular domain, PD-1 or CTLA-4. Co-transduction with a regular CAR leads to the activation of the iCAR, which can inhibit the activity of the co-expressed CAR, thereby limiting the undesired CAR activation[92]. Other improved CAR designs include tandem CARs (TanCAR)[79] and switchable CARs[93], which specifically expand the range of TAAs that can be simultaneously targeted. Bispecific CAR-T cells are designed by engineering a single CAR molecule with two or more distinct binding domains. Multi-target CAR-T cell therapies are produced by mixing different CAR-T cell products targeting single antigens before infusion or transducing T cells with multiple CAR constructs CAR-T cells expressing bispecific T cell engagers (BiTEs) recruit bystander T cells against a second TAA[94]. The synNotch CAR-T cells offer benefits beyond enhanced tumor specificity by preventing premature T cell differentiation and exhaustion, further enabling T cells to maintain a long-lived memory[45,85]. Figure 2 shows the schematic representation of all the novel CARs designs discussed in this review.
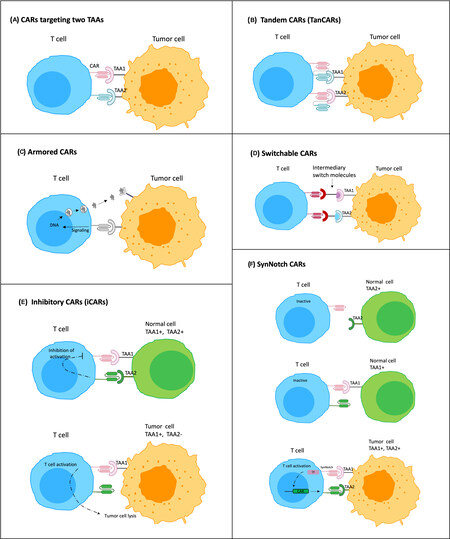
Figure 2. Schematic of novel CARs designed to enhance the efficacy of CAR-T cells in solid tumors. (A) CARs targeting two TAAs (dual CAR expressing T cell); (B) tandem CARs (TanCARs) that have two different scFVs connected in tandem to one CAR T-cell and deal with antigen escape or multiple tumor antigens; (C) armored CARs secreting cytokines, antibodies and other immunomodulatory agents in the TME upon CAR-antigen engagement; (D) switchable CARs that utilize different target molecules specific for each TAA and can redirect CAR-T cells against various types of tumors; (E) inhibitory CARs that, upon interaction with normal cells expressing the TAA, prevent downstream signaling cascades inhibiting CAR-T cell function; (F) synNotch CARs, in which recognition of a specific TAA (TAA1) on tumor cell through synNotch receptor, cleaves the receptor to release a transcription factor (TF) specific for the induction and expression of a CAR against a second TAA (TAA2).
Combination of CAR-T cells and checkpoint inhibitors
Limited engraftment and persistence of CAR-T cells are generally observed in solid tumors post infusion. One of the ways to ease this is by regional administration of CAR-T cells, where possible. In a recent MPM study, local, intrapleural delivery of MSNL-targeted CAR-T cells, followed by administration of Pembrolizumab, demonstrated promising survival outcomes, with a median OS of 23.9 months, despite including patients with mesothelin-positive MPM as low as 10%[81]. This study highlights the potential of CAR-T cell therapy in solid tumors such as MPM. CAR-T cells’ precise antitumor immune response can be further amplified by the addition of ICI agents that counteract the immune suppressive TME that undermines optimal CAR-T cell efficacy. A review paper by Chintal et al. on MPM and other solid tumors explores the advantage of combination immunotherapy strategies to enhance endogenous and adoptively transferred immunity[95]. In a recent paper, Raghav et al. investigated two drugs, PD-L1 (atezolizumab) and vascular endothelial growth factor (VEGF) (bevacizumab) blockade, in 20 patients with advanced MPeM either after progression or intolerance to prior pemetrexed based chemotherapy[29]. Efficacy of AtezoBevo in relation to response rates and survival in advanced MPeM previously treated with chemotherapy surpassed outcomes with a median progression-free survival (PFS) of 17.6 months, and one-year OS of 85%. Further, MPeM biomarker analysis revealed epithelial-mesenchymal transition phenotype which was associated with therapeutic resistance to both AtezoBevo combination and prior to platinum-pemetrexed chemotherapy, even among patients with epithelioid subtype[29]. Combination immunotherapies comprising CAR-T cells and ICI agents are currently under clinical trial investigation.
Induction of bystander effects following epitope spreading
CAR-T cell therapy for solid tumors, in which the targeted antigen is not expressed on all of the tumor cells, could be more successful when combined with immunomodulatory agents that enhance bystander effects. Bystander effects, in the context of CAR-T cells, refer collectively to the effects induced as a consequence of tumor cell lysis by CAR-T cells on the tumor cells that do not express the CAR target antigen/s[96,97]. These effects include the direct tumor cell lysis from cytokines secreted as a byproduct from CAR-T cells or tumor cell lysis caused by immune cells attracted to the tumor by these secreted cytokines. However, it is thought that the most potent bystander effects are due to epitope spreading. Following CAR-T cell tumor cell lysis, release/spread tumor debris containing tumor epitopes are released/spread in the TME and taken up by antigen-presenting cells (APCs). Cross-presentation of these epitopes to lymphocytes in the lymph nodes ultimately generates an endogenous T cell response against tumor antigens not originally targeted by the CAR-T cells[96]. There is evidence that tumor-specific CD8+ T cells can mediate this process of antigen or epitope spreading via (i) therapeutic cancer vaccines; (ii) immune checkpoint inhibition; and (iii) adoptive T cell therapies[82]. During a clinical trial of MSLN-targeting CAR-T cell therapy in patients with MSLN-expressing solid malignancies, two patients (one with advanced MPM and the other with metastatic pancreatic cancer) demonstrated clinical evidence of broad antitumor immune response consistent with epitope spreading[83]. Following treatment with MSLN CAR-T cells, new anti-self antibodies were seen in these two patients[83], who presented with clinical antitumor activity despite not receiving lymphodepletion therapy before the CAR-T cell infusion[83]. Another study by the same group highlighted that clonal expansion of endogenous T cells could be induced by anti-MSLN CAR-T cells in several patients with advanced solid cancers, which was detected by deep sequencing of the TCR beta chain[98]. This was not observed in patients receiving lymphodepletion before CAR-T cell transfer[98]. These findings suggest that CAR-T cells elicit a ‘vaccine’ effect with potential therapeutic implications. Additionally, CAR-T cells can induce augmentation of humoral responses, as well as epitope spreading in patients, both of which appear to be hampered by lymphodepletion. An example of epitope spreading has also been reported with bispecific antibody (BiAb) therapy. A bispecific T cell engager (BiTE) antibody targeting Wilms’ tumor protein (WT1) led to the expansion of secondary T cell clones with specificity for TAAs other than WT1 in vitro co-cultures of patient PBMCs with autologous tumor cells[80]. This study indicates that the therapeutic efficacy of BiTEs could be attributed partly to epitope spreading following intracellular tumor antigen targeting.
One of the few preclinical studies on the immunologic bystander effects of CAR-T cell therapy so far was conducted on syngeneic animal models of MM[84]. In this study, mesothelin-targeting CAR-T cells were unable to eliminate tumors that were < 100% positive for mesothelin, demonstrating the limitation of CAR-T cell therapy when faced with antigen-heterogenous tumors. The authors tried to augment therapeutic efficacy by inducing bystander effects in various ways and found that pre-treatment of mice with a low, non-lymphodepleting dose of cyclophosphamide (CTX), followed by CAR-T cell therapy, had a curative effect in tumors that were < 100% mesothelin positive[84]. This effect was demonstrated to be dependent on endogenous CD8 T cells but not on basic leucine zipper transcription factor ATF-like 3 (BATF3)-dependent type 1 dendritic cells (DCs)[84]. In another study by Lai et al., T cells were engineered to secrete the dendritic cell (DC) growth factor Fms-like tyrosine kinase 3 ligand (FLT3L). FLT3L-secreting T cells expanded intratumoral conventional type 1 dendritic cells (DCs) and substantially increased host DC and T cell activation when combined with immune agonists poly (I:C) and anti-4-1BB[99]. These data reveal that augmenting endogenous DCs is a promising strategy to overcome the clinical problem of antigen-negative tumor escape following adoptively-transferred cell therapy[99]. A study using a syngeneic mouse melanoma model with defined tumor antigens showed that epitope spreading after treatment with TCR-engineered T cells targeted to the melanoma tumor antigen gp100 was minimal[100]. However, epitope spreading mediated by BATF3-dependent DCs and bystander effects was seen following co-transduction of T cells with a second TCR that recognizes a bacterially-expressed antigen (ovalbumin) and treating tumors with modified Listeria bacteria that expresses ovalbumin[100].
CONCLUSION
CAR-T cell therapy for MM presents unique biological and clinical challenges similar to other advanced solid malignancies. One of these challenges is tumor-antigen heterogeneity, which allows antigen-negative tumor cells to escape CAR-T cell lysis and continue to grow. Theoretically, universal antigen expression on tumors should not be required for CAR-T cells to eliminate the tumor, as CAR-T cell function can induce an immunologic bystander effect, resulting in the non-antigen-specific antitumor immune response. The latter is aided either by innate immune cell populations with tumor lytic abilities or, most significantly, by cross-presentation of a diverse antigen repertoire to CD4+ and CD8+ T cells by antigen-presenting cells (APCs) in the lymph nodes, resulting in activation of the T cell cytotoxic functions.
In the few studies conducted so far, it is clear that CAR-T cells are unable to induce strong bystander effects in solid tumor models, yet some promising strategies exist that can potentiate a broad endogenous antitumor response. Combination of CAR-T cells with immunomodulatory agents, and the design of novel, sophisticated CAR constructs with secretory abilities might be ways to manage TME’s immunosuppression, allowing CAR-T cells and the endogenous T cell repertoire to function more efficiently. While the development of more advanced CAR-T cell therapies is underway, the presence or lack of bystander effects following CAR-T cell therapy needs to be thoroughly investigated in more preclinical studies and, more importantly, clinical trials that will ultimately determine whether these therapies can exhibit their full therapeutic potential in solid tumors like MM.
DECLARATIONS
Authors’ contributionsEqually contributed to the design, writing, critically reviewing and final approval of the manuscript: D'Souza RR, Dimou P, Bughda R, Hawkins E, Laboreiro Babe C, Klampatsa A
Availability of data and materialsNot applicable.
Financial support and sponsorshipKlampatsa A is supported by the Cris Cancer Foundation.
Conflict of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Brims F. Epidemiology and clinical aspects of malignant pleural mesothelioma. Cancers (Basel) 2021;13:4194.
2. Ziółkowska B, Cybulska-Stopa B, Papantoniou D, Suwiński R. Systemic treatment in patients with malignant pleural mesothelioma - real life experience. BMC Cancer 2022;22:432.
3. Espinoza-Mercado F, Borgella JD, Berz D, et al. Disparities in compliance with national guidelines for the treatment of malignant pleural mesothelioma. Ann Thorac Surg 2019;108:889-96.
4. Baas P, Scherpereel A, Nowak AK, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet 2021;397:375-86.
5. Broeckx G, Pauwels P. Malignant peritoneal mesothelioma: a review. Transl Lung Cancer Res 2018;7:537-42.
6. Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 2003;21:2636-44.
7. Kindler HL, Ismaila N, Armato SG, et al. Treatment of malignant pleural mesothelioma: American society of clinical oncology clinical practice guideline. J Clin Oncol 2018;36:1343-73.
8. Zalcman G, Mazieres J, Margery J, et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open-label, phase 3 trial. Lancet 2016;387:1405-14.
9. Neumann V, Günther S, Müller KM, Fischer M. Malignant mesothelioma - German mesothelioma register 1987-1999. Int Arch Occup Environ Health 2001;74:383-95.
10. Tracey EKT, Dobrovic A, Currow D. Cancer in NSW: incidence and mortality report 2008. Sydney: Cancer Institute NSW, 2010.
11. Tano ZE, Chintala NK, Li X, Adusumilli PS. Novel immunotherapy clinical trials in malignant pleural mesothelioma. Ann Transl Med 2017;5:245.
12. Rijavec E, Biello F, Barletta G, Dellepiane C, Genova C. Novel approaches for the treatment of unresectable malignant pleural mesothelioma: a focus on immunotherapy and target therapy (Review). Mol Clin Oncol 2022;16:89.
13. Chen N, Li X, Chintala NK, Tano ZE, Adusumilli PS. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr Opin Immunol 2018;51:103-10.
14. Albelda SM. Tumor antigen heterogeneity: the “Elephant in the Room” of adoptive T-cell therapy for solid tumors. Cancer Immunol Res 2020;8:2.
15. Garland LL, Rankin C, Gandara DR, et al. Phase II study of erlotinib in patients with malignant pleural mesothelioma: a Southwest Oncology Group Study. J Clin Oncol 2007;25:2406-13.
16. Govindan R, Kratzke RA, Herndon JE, et al. Gefitinib in patients with malignant mesothelioma: a phase II study by the Cancer and Leukemia Group B. Clin Cancer Res 2005;11:2300-4.
17. Ou WB, Hubert C, Corson JM, et al. Targeted inhibition of multiple receptor tyrosine kinases in mesothelioma. Neoplasia 2011;13:12-22.
18. Ou SH, Moon J, Garland LL, et al. SWOG S0722: phase II study of mTOR inhibitor everolimus (RAD001) in advanced malignant pleural mesothelioma (MPM). J Thorac Oncol 2015;10:387-91.
19. Avelumab in metastatic or locally advanced solid tumors (JAVELIN solid tumor). Available from: https://ClinicalTrials.gov/show/NCT01772004 [Last accessed on 21 July 2022].
20. Gounant V, Brosseau S, Zalcman G. Immunotherapy, the promise for present and future of malignant pleural mesothelioma (MPM) treatment. Ther Adv Med Oncol 2021;13:17588359211061956.
21. Brockwell NK, Alamgeer M, Kumar B, Rivalland G, John T, Parker BS. Preliminary study highlights the potential of immune checkpoint inhibitors in sarcomatoid mesothelioma. Transl Lung Cancer Res 2020;9:639-45.
22. Zalcman G, Mazieres J, Greillier L, et al. Second/third-line nivolumab vs nivo plus ipilimumab in malignant pleural mesothelioma: long-term results of IFCT-1501 MAPS2 phase IIR trial with a focus on hyperprogression (HPD) Ann Oncol 2019. p. v747.
23. Pagano M, Ceresoli GL, Zucali PA, et al. Randomized phase II study on gemcitabine with or without ramucirumab as second-line treatment for advanced malignant pleural mesothelioma (MPM): Results of Italian Rames Study JCO 2020. p. 9004.
24. Toyokawa G, Takenoyama M, Hirai F, et al. Gemcitabine and vinorelbine as second-line or beyond treatment in patients with malignant pleural mesothelioma pretreated with platinum plus pemetrexed chemotherapy. Int J Clin Oncol 2014;19:601-6.
25. Tourkantonis I, Makrilia N, Ralli M, et al. Phase II study of gemcitabine plus docetaxel as second-line treatment in malignant pleural mesothelioma: a single institution study. Am J Clin Oncol 2011;34:38-42.
26. Kim RY, Li Y, Marmarelis ME, Vachani A. Comparative effectiveness of second-line immune checkpoint inhibitor therapy versus chemotherapy for malignant pleural mesothelioma. Lung Cancer 2021;159:107-10.
27. Popat S, Curioni-Fontecedro A, Dafni U, et al. A multicentre randomised phase III trial comparing pembrolizumab versus single-agent chemotherapy for advanced pre-treated malignant pleural mesothelioma: the European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann Oncol 2020;31:1734-45.
28. Yarchoan M, Albacker LA, Hopkins AC, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019;4:126908.
29. Raghav K, Liu S, Overman MJ, et al. Efficacy, safety, and biomarker analysis of combined PD-L1 (Atezolizumab) and VEGF (Bevacizumab) blockade in advanced malignant peritoneal mesothelioma. Cancer Discov 2021;11:2738-47.
30. Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L. Trial watch: adoptively transferred cells for anticancer immunotherapy. Oncoimmunology 2017;6:e1363139.
32. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov 2013;3:388-98.
33. Thistlethwaite FC, Gilham DE, Guest RD, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother 2017;66:1425-36.
34. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther 2010;18:413-20.
35. Chang K, Pai LH, Batra JK, Pastan I, Willingham MC. Characterization of the antigen (CAK1) recognized by monoclonal antibody K1 present on ovarian cancers and normal mesothelium. Cancer Res 1992;52:181-6.
36. Pastan I, Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res 2014;74:2907-12.
37. Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov 2016;6:133-46.
38. Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol 2000;20:2902-6.
39. Castelletti L, Yeo D, van Zandwijk N, Rasko JEJ. Anti-mesothelin CAR T cell therapy for malignant mesothelioma. Biomark Res 2021;9:11.
40. Klampatsa A, Dimou V, Albelda SM. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin Biol Ther 2021;21:473-86.
41. Klampatsa A, Achkova DY, Davies DM, et al. Intracavitary “T4 immunotherapy” of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett 2017;393:52-9.
42. Al-Taei S, Salimu J, Lester JF, et al. Overexpression and potential targeting of the oncofoetal antigen 5T4 in malignant pleural mesothelioma. Lung Cancer 2012;77:312-8.
43. Klampatsa A, Haas AR, Moon EK, Albelda SM. Chimeric antigen receptor (CAR) T cell therapy for malignant pleural mesothelioma (MPM). Cancers (Basel) 2017;9:115.
44. Thayaparan T, Petrovic RM, Achkova DY, et al. CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. Oncoimmunology 2017;6:e1363137.
45. Hyrenius-Wittsten A, Su Y, Park M, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med 2021;13:eabd8836.
46. Bughda R, Dimou P, D’Souza RR, Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: launching an attack on tumor stroma. Immunotargets Ther 2021;10:313-23.
47. Petrausch U, Schuberth PC, Hagedorn C, et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer 2012;12:615.
48. Schuberth PC, Hagedorn C, Jensen SM, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med 2013;11:187.
49. Hiltbrunner S, Britschgi C, Schuberth P, et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann Oncol 2021;32:120-1.
50. Blum Y, Meiller C, Quetel L, et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat Commun 2019;10:1333.
51. Yoshikawa Y, Sato A, Tsujimura T, et al. Frequent inactivation of the BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci 2012;103:868-74.
52. Kiyotani K, Park JH, Inoue H, et al. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology 2017;6:e1278330.
53. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013;501:346-54.
54. Oehl K, Vrugt B, Opitz I, Meerang M. Heterogeneity in malignant pleural mesothelioma. Int J Mol Sci 2018;19:1603.
55. Lee HS, Jang HJ, Choi JM, et al. Comprehensive immunoproteogenomic analyses of malignant pleural mesothelioma. JCI Insight 2018;3:98575.
56. Chen R, Lee WC, Fujimoto J, et al. Evolution of genomic and T-cell repertoire heterogeneity of malignant pleural mesothelioma under dasatinib treatment. Clin Cancer Res 2020;26:5477-86.
57. Sneddon S, Rive CM, Ma S, et al. Identification of a CD8+ T-cell response to a predicted neoantigen in malignant mesothelioma. Oncoimmunology 2020;9:1684713.
58. Mansfield AS, Peikert T, Smadbeck JB, et al. Neoantigenic potential of complex chromosomal rearrangements in mesothelioma. J Thorac Oncol 2019;14:276-87.
59. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov 2018;8:1219-26.
60. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 2020;17:147-67.
61. Minnema-Luiting J, Vroman H, Aerts J, Cornelissen R. Heterogeneity in immune cell content in malignant pleural mesothelioma. Int J Mol Sci 2018;19:1041.
62. Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med 2017;68:139-52.
64. Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature 1995;375:148-51.
65. Alvarez-Vallina L, Russell SJ. Efficient discrimination between different densities of target antigen by tetracycline-regulatable T bodies. Hum Gene Ther 1999;10:559-63.
66. Watanabe K, Terakura S, Martens AC, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J Immunol 2015;194:911-20.
67. Chmielewski M, Hombach AA, Abken H. CD28 cosignalling does not affect the activation threshold in a chimeric antigen receptor-redirected T-cell attack. Gene Ther 2011;18:62-72.
68. Frigault MJ, Lee J, Basil MC, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res 2015;3:356-67.
69. Viganò S, Utzschneider DT, Perreau M, Pantaleo G, Zehn D, Harari A. Functional avidity: a measure to predict the efficacy of effector T cells? Clin Dev Immunol 2012;2012:153863.
70. Zeh HJ, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol 1999;162:989-94.
71. Walker AJ, Majzner RG, Zhang L, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther 2017;25:2189-201.
72. Arcangeli S, Rotiroti MC, Bardelli M, et al. Balance of Anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol Ther 2017;25:1933-45.
73. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18:843-51.
74. Liu X, Jiang S, Fang C, et al. Affinity-Tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 2015;75:3596-607.
75. Drent E, Themeli M, Poels R, et al. A rational strategy for reducing On-Target Off-Tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther 2017;25:1946-58.
76. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9:eaaa0984.
78. Larson RC, Kann MC, Bailey SR, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature 2022;604:563-70.
79. Hegde M, Mukherjee M, Grada Z, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest 2016;126:3036-52.
80. Dao T, Pankov D, Scott A, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol 2015;33:1079-86.
81. Adusumilli PS, Zauderer MG, Rivière I, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the Anti-PD-1 agent pembrolizumab. Cancer Discov 2021;11:2748-63.
82. Brossart P. The Role of antigen spreading in the efficacy of immunotherapies. Clin Cancer Res 2020;26:4442-7.
83. Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014;2:112-20.
84. Klampatsa A, Leibowitz MS, Sun J, Liousia M, Arguiri E, Albelda SM. Analysis and augmentation of the immunologic bystander effects of car T cell therapy in a syngeneic mouse cancer model. Mol Ther Oncolytics 2020;18:360-71.
85. Choe JH, Watchmaker PB, Simic MS, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med 2021;13:eabe7378.
86. Anurathapan U, Chan RC, Hindi HF, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther 2014;22:623-33.
87. Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol 2016;9:56.
88. Smith TT, Moffett HF, Stephan SB, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest 2017;127:2176-91.
89. Chen SH, Hung WC, Wang P, Paul C, Konstantopoulos K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci Rep 2013;3:1870.
90. Zhang L, Yu Z, Muranski P, et al. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther 2013;20:575-80.
91. Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 2016;126:3130-44.
92. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 2013;5:215ra172.
93. Rodgers DT, Mazagova M, Hampton EN, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci USA 2016;113:E459-68.
94. Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 2019;37:1049-58.
95. Chintala NK, Restle D, Quach H, et al. CAR T-cell therapy for pleural mesothelioma: Rationale, preclinical development, and clinical trials. Lung Cancer 2021;157:48-59.
96. Sánchez-Paulete AR, Teijeira A, Cueto FJ, et al. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol 2017;28:xii44-55.
97. Gulley JL, Madan RA, Pachynski R, et al. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J Natl Cancer Inst 2017:109.
98. Kim RH, Plesa G, Gladney W, et al. Effect of chimeric antigen receptor (CAR) T cells on clonal expansion of endogenous non-CAR T cells in patients (pts) with advanced solid cancer. JCO 2017;35:3011-3011.
99. Lai J, Mardiana S, House IG, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol 2020;21:914-26.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
D'Souza RR, Dimou P, Bughda R, Hawkins E, Leboreiro Babe C, Klampatsa A. Overcoming tumor antigen heterogeneity in
AMA Style
D'Souza RR, Dimou P, Bughda R, Hawkins E, Leboreiro Babe C, Klampatsa A. Overcoming tumor antigen heterogeneity in
Chicago/Turabian Style
D'Souza, Reena R., Paraskevi Dimou, Reyisa Bughda, Elizabeth Hawkins, Clara Leboreiro Babe, Astero Klampatsa. 2022. "Overcoming tumor antigen heterogeneity in
ACS Style
D'Souza, RR.; Dimou P.; Bughda R.; Hawkins E.; Leboreiro Babe C.; Klampatsa A. Overcoming tumor antigen heterogeneity in
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 30 clicks
Cite This Article 30 clicks

 Like This Article 26
likes
Like This Article 26
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.