
Pathways linking Alzheimer's disease risk genes expressed highly in microglia

Abstract
Microglia in the brain are exquisitely vigilant to their surroundings. They are dispersed throughout the brain parenchyma where they continually receive and integrate large numbers of incoming signals. They become activated once a tightly controlled signalling threshold is reached. This can lead to a cascade of cellular and molecular changes culminating in the recognition and engulfment of self and non-self structures ranging from macromolecules to whole cells depending on the initiating signal. Once internalised, they digest and where appropriate, present antigens to aid future recognition of pathogens. Their response to pathogenic signals in diseases such as Alzheimer’s disease (AD) has long been recognised, but recent genetic findings have cemented their direct causal contribution to AD and thus the potential to target them or their effector pathways as a possible treatment strategy. Around 25% of the ~84 AD risk genes have enriched or exclusive expression in microglia and/or are linked to immune function*. Ongoing work suggests many of these genes connect within important microglial molecular networks as ligand activators (IL34), immune receptors (TREM2, MS4A4A, HLA-DQA1 & CD33), signalling intermediates (PLCG2, PTK2B & INPP5D) or effector mechanisms (ABI3 & EPHA1). In some cases, evidence links them to specific core pathogenic immune responses and cell mechanisms such as complement (CR1 & CLU) or cytoskeletal machinery (ABI3, EPHA1 and FERMT2). However, more work is needed to establish whether these risk variants lead to gain or loss of protein function and to connect them to other genes within effector pathways and downstream cell processes which themselves could be tractable targets for treatment development. Brain tissue analysis and cell models of genetic risk carriers will help enormously to expand the knowledge of pathways and cell processes important to AD. It will also help direct efforts to develop suitable agonist or antagonist tool compounds for testing target tractability for therapeutic development and for establishing appropriate biomarkers to evaluate efficacy in AD. Ultimately, it should pave the way for developing treatments for AD and potentially other diseases where boosting or dampening key microglia pathways could significantly impact disease outcomes irrespective of genetic status.
Keywords
Introduction
Microglia have a finely tuned array of receptors that enable them to discriminate incoming homeostatic, damage and pathogenic signals and respond appropriately. In Alzheimer’s disease (AD), microglia are exposed to a complex array of damage and/or pathogenic signals which challenge their capacity to respond adequately or appropriately. AD is a slow progressive neurodegenerative disease characterised not only by a characteristic pattern of neuronal cell loss in the brain, but also by extracellular protein aggregates composed of Aβ (processed from APP) depositing as plaques and intracellular protein aggregates composed of hyperphosphorylated Tau (MAPT) depositing as neurofibrillary tangles. This pathological sequelae is the same in people with sporadic disease where the cause is unknown and in patients developing AD who also have a risk variant in one or more AD risk genes. We have collated a list of 84 AD risk genes of which 20 have highly enriched or exclusive expression in microglia. Many of these genes can be linked to common signalling pathways, particularly converging on the cytoskeletal system, and thus along with other pathways are expected to have important consequences for the coordinated phagocytic response of microglia in the AD brain. We discuss the various roles for microglia in the brain, including their normal function and behaviour and when presented with different pathogenic and damage signals in AD. We review the current knowledge of AD risk genes and the variants impacting their function. We establish which of these genes are likely to have causal effects in microglia and connect AD risk genes to signalling pathways in microglia. This provides a framework by which further work can test and evaluate these pathways and identify tractable targets to guide therapeutic development.
The brain immune system
Unlike other tissues in the body, the blood brain barrier is tightly regulated, leading to a largely localised and autonomous immune system. Resident microglia are the predominant immune cell present in the brain. They provide innate immune defence against infection and trauma, and maintain healthy tissue and homeostasis by clearing apoptotic cells and performing synaptic pruning. Although the brain is a highly vascularised organ, the blood brain barrier prevents the adaptive immune system from having a significant role in the brain (discussed below). There is evidence however that microglia have the capacity for immune memory from prior exposure to the same activating signals, leading to an enhanced (training) or diminished (tolerance) response to the same signal during subsequent exposures[1].
The extent to which peripheral immune cells can enter or leave the brain parenchyma is controversial. The three main barriers controlling entry (or exit) of cells and solutes are (1) the blood-brain barrier (BBB, consisting of a neurovascular unit containing endothelial cells with connecting tight junctions, pericytes and astrocytes); (2) the blood-CSF barrier (BCSFB, consisting of either epithelial cells and their joining tight junctions within the choroid plexuses or the arachnoid and dura mater surrounding microvessels, arteries, veins and lymph vessels at the brain surface); and (3) the CSF-brain barrier (consisting of ependymal cells at the ventricular surface and the pial-glial barrier of the outer surface of the brain parenchyma)[2]. Despite these barriers, there are still many examples of their breach in autoimmune diseases such as Multiple Sclerosis[3], Systemic Lupus Erythematosus[4,5], primary and metastatic brain tumours[6,7], infections[8] and in advanced stages of diseases such as AD[9-11]. Under normal homeostatic conditions, there is negligible cell movement between these compartments, although the selective influx/efflux of proteins and other molecules can occur particularly in early life[12]. However, the view that the brain is immune privileged is no longer considered true. Evidence shows antigens and antigen presenting cells originating in the brain parenchyma can reach the dural and cervical lymphatics[2,13,14], and thereby communicate with the deep cervical lymph nodes[15]. Via the blood they can then illicit adaptive and humoral responses which can return full circle to have effector function in the brain[16]. Whilst the adaptive arm of the immune system has less evidence for direct activity in the brain parenchyma, it has long been recognised that the innate immune system plays a central role in brain health and disease. The main cell type mediating this is the microglial cell. Microglia emerge very early in development in the yolk sac[17,18] where they expand to populate ~5%-12% of the total brain cell population[19].
Long-lived homeostatic microglia[20-22] reside alone in distinct territories as ramified cells under normal circumstances. The cells survey their immediate surroundings for damage or pathogenic signals in the brain parenchyma by extending and retracting their cell membranes to form specialised highly motile filopodia protrusions. Once receptors on the filopodia (such as purinergic receptors) engage an external or internal signal (such as ATP), microglia typically respond by retracting and repurposing their membrane protrusions. Either through direct contact or migration following detection of a diffusible signal, they surround, engulf and digest the pathogen or damaged tissue, cells or cell fragments thus potentially remodelling tissue or cell activity in the immediate vicinity. Depending on the signal, they may also generate toxic cytokine or chemokine signals which attract other microglia and in so doing can form nodules of microglia clusters. Activation of so-called inflammatory or anti-inflammatory cascades in microglia can involve engagement of Fcγ receptors, complement receptors, toll-like receptors (TLR), RIG-like receptors (RLRs), Aim-2-like receptors (ALRs), C-type lectins, advanced glycosylation end product receptor (RAGE), scavenger receptors, N-formyl peptide receptors (FPRs) or NOD-like receptors (NLRs, particularly NALP3) or TREM2 either working alone or together with other neurotransmitter receptors expressed on microglia[23-26].
In addition to degradation through the lysosomal pathway, phagocytic/endosomal engulfment can lead to the presentation of antigens through the MHC Class II system which can initiate the adaptive and humoral systems either locally within the brain or through the systemic immune system. Other minor populations of immune cells can be detected in the brain under homeostatic conditions. These lymphocytes, macrophages and dendritic cells usually reside in the margins of brain parenchyma or in specific niches such as the meninges, but nevertheless under the right conditions can play important roles in health and disease[27-29].
Microglia impact normal behaviour and development
Beyond their role in combatting pathogens and responding to damage, microglia are increasingly recognised for their important role in shaping behaviour. They can have direct effects on neuronal cell function during normal development and respond to external experience. There is much evidence to demonstrate microglia make activity-dependent contact with neuronal cells[30-35], impacting the output of local neural circuits[36,37]. But more than this, they are able to engulf synaptic components[38-41] and they can “nibble” the membranes of presynaptic neurons at the peak period of brain remodelling in early development, altering the structure and composition of these membranes and impacting postsynaptic spine head filopodia[42]. Others have shown that microglia respond to IL-33 produced by stimulated neuronal cells by remodelling the extracellular perineuronal matrix which in turn affects dendritic spine and synaptic density and behaviours associated with learning and memory[43]. Not only do these activities occur during developmental remodelling, but also throughout life in response to experience[44,45]. Synaptic remodelling involving microglia is commonly found to be dependent on the activation of the complement cascade[39,41,46-48], although not all phagocytic activities of microglia have been shown to rely on a complement-mediated mechanism. Microglia can also associate with axon tracts in white matter undergoing myelination during early postnatal development and phagocytose oligodendrocytes or their precursors, even in the absence of overt apoptotic signals[49,50]. Recently, microglia were found to directly phagocytose excessive and ectopic myelin during this period, presumably an additional function for shaping neural circuits and behaviour[51]. The molecular signals facilitating this have still to be elucidated. Many of the activities described above are particularly prominent in adolescence where they appear to coincide with the peak of neuronal plasticity, which incidentally is when TREM2 is most active (discussed below).
Microglia respond to damage in Alzheimer’s disease
Microglia can efficiently detect and clear cellular and acellular material following their detection. This includes acute damage caused by trauma or ongoing damage which emerges in chronic diseases such as AD. In the case of AD, there are a myriad of possible “competing” damage related signals simultaneously arising from amyloid plaques, fibrils and insoluble Aβ aggregates, damaged myelin, extracellular matrix and stressed/degenerating neurons. Important challenges for AD therapeutics are reconciling which microglia responses are protective and which are detrimental, identifying the most impactful pathways to target for drug development, identifying the best treatment window and targeting pathways unique to microglia to minimise off-target effects. This is a big challenge. Many receptors in microglia are promiscuous, activated by a myriad of damage and pathogen associated signals[52]. Further functional complexity arises through receptor clustering and dimerisation. TLR4, widely found on myeloid cells, can be activated by the bacterial wall component lipopolysaccharide (LPS), hyaluronan, biglycan, heparan sulphate and CD36 associated Aβ[53]. TREM2 is microglia-specific, but appears able to bind a number of ligands including Aβ[54,55], apolipoproteins such as APOE[56,57], anionic and zwitterionic lipids[58] and anionic carbohydrates[59]. RAGE (AGER) which binds Aβ, S100A12 and S100B is expressed by a number of brain cell types including microglia, neurons and endothelial cells[60] where it is considered an important receptor for regulating amyloid angiopathy[61].
As diseases such as AD develop, microglia can lose their solitary behaviour and form clusters around amyloid plaques either through local proliferation or migration[62-70]. The morphology of microglia become more amoeboid as they pivot towards a so-called reactive profile, altering the repertoire of proteins they express in favour of those needed to mount an immune response. Central to this is phagocytosis[71-74]. Some suggest that the purpose of the clustering behaviour of microglia is a form of amyloid containment or “corralling” to neutralise its toxic effects, rather than for the purpose of amyloid removal[75-77]. Certainly, the capacity for aged microglia to phagocytose amyloid is considered by some to be very poor in AD[78-82] and microglia may even propagate rather than reduce amyloid deposition via NLRP3-ASC speck seeding[83]. This may explain why amyloid accumulates in AD[84]. However, many others have shown that microglia under the right conditions can very efficiently engulf and remove amyloid. For example, following amyloid immunotherapy[68,85-92]. Microglia propagation has also been suggested for Tau aggregates either through microglia engulfment of neuronal cells containing Tau aggregates or via Tau aggregates released at synapses which are subsequently repackaged and released by microglia within exosomes[93-96]. Because of the unique combination of genes expressed by amyloid-associated microglia, the term “DAM” microglia (Damage-Associated Microglia) has been coined to describe a relatively consistent expression profile that marks the activation journey of these cells in mouse models of AD[71-74,97-99]. This phenotype is broadly characterised by high levels of expression of genes involved in complement, antigen presentation, actin regulation and ribosomes consistent with immune activation and phagocytic function. TREM2 (discussed below) occupies a central position in the DAM cell expression profile and when absent, causes a rippling impact on other brain cell types associated with AD, notably oligodendrocytes and endothelial cells[73,99,100]. A broadly similar expression profile has been observed in “PAM” (Proliferative-region-Associated Microglia) in white matter during normal early development[49], suggesting there is some conservation of function in reactive microglia across different brain contexts. However, there may be important species differences. Recently, the mouse DAM profile was reported to not have strong overlap with human “HAM” (Human Alzheimer’s Microglia) profiles from superior frontal gyrus generated from bulk single-sorted ITGAM (CD11b) expressing cells from late-stage pathology cases[101].
Fibrillar Aβ appears to interact with CD36, AGER and TREM2 receptors expressed by microglia. In the case of CD36, binding Aβ and additional interactions with TLR4:TLR6 triggers an inflammatory response. This leads to NFKB1 (NF-κβ)-dependent production of CXCL1, CXCL2 and CCL9 cytokines, via the MYD88 signalling pathway, CCL5 cytokine production via the TICAM1 signalling pathway and IL1β secretion, through the priming and activation of the NLRP3 inflammasome. Activation of the NLRP3 inflammasome leads to the release of ASC specks[102-104]. As mentioned earlier, ASC specks can bind extracellular Aβ and facilitate further aggregate seeding[83], thus perpetuating a continuous cycle of pathology. NLRP3-ASC mediated activity also exacerbates Tau hyperphosphorylation and aggregation[105]. Interaction of fibrillar Aβ with AGER leads to activation of NF-κb, phosphoinositide 3-kinase (PI3K), Janus kinase, signal transducer and activator of transcription and mitogen-activated protein kinase (MAPK) signalling pathways, which leads to induction of pro-inflammatory cytokines such as TNF[53,106]. In contrast, activation of TREM2-TYROBP (DAP12) is considered to largely occur without the production of pro-inflammatory molecules and is thus considered beneficial for AD. TREM2-TYROBP signalling triggers many protein and lipid phosphorylation cascades causing calcium mobilization, integrin activation, mTOR and MAPK signalling, activation of energetic metabolism[107,108] and cytoskeleton activation via SYK[109]. Many of these pathways culminate in cytoskeletal changes that help drive phagocytosis[110]. Conversely, loss of TREM2 leads to deficits in phagocytosis, at least in the uptake of Aβ, myelin debris, apoptotic neurons and/or E.coli[111-115] and an inability to switch to a glycolytic state[108].
The impact of TREM2 in AD appears to be determined by the pathogenic and temporal context. Over-expression, activation or restoration of normal TREM2 function has been shown to have largely beneficial outcomes in amyloid-driven mouse models[116-119] while knock-out or haploinsufficiency exacerbates amyloid-mediated pathologies[76,120-123]; although some studies are at variance with this[124]. In Tau-driven models, the picture is more complex with some studies showing loss of TREM2 to be protective in the presence of Tau pathology[124,125] while others show it leads to worse outcomes[126,127]. These differences may lie in the timing and/or severity of Tau pathology. TREM2 knock-out or bi-allelic hypomorphic variant mouse models (modelling Nasu-Hakola disease) by themselves do not lead to amyloid or Tau pathology, but in keeping with a protective role for TREM2, these mice exhibit greater pro-inflammatory markers, synaptic dysfunction, neuronal cell loss and worse cognition with age[128,129] and mirror the symptoms described for Nasu-Hakola disease[130]. Thus knock-out models of TREM2 recapitulate the more extreme variants rather than the subtle rs75932628, R47H heterozygous variant linked to AD[108,112].
Complement-mediated remodelling of synapses has long been recognised as an important driver of normal brain plasticity and can significantly impact memory performance[39,41]. The complement cascade has also been shown to be activated in AD mouse model brains mirroring areas of pathology[48]. Activation of C1q (C1QA, B and C) within the C1 complement complex leads to opsonisation or tagging of material which can then activate Fcγ receptor-dependent or -independent pathways in microglia. The Fcγ receptor-independent C1q pathway signals through complement receptor 3 (CR3; ITGAM/ITGB2) while C1q-IgG mediated signalling is through complement receptor 1 (CR1) and Fcγ receptor pathways. Loss of protective sialic acid residues from glycoproteins on cell surfaces such as neuronal cells by neuraminidase activity can also lead to their tagging by C1q and subsequent CR3 activation resulting in neuronal phagocytosis[39,131,132]. Alternatively, desialylated glycoprotein-C1q-calreticulin binding on neurons activates the microglia LRP1 receptor, also leading to phagocytosis[133]. It is noteworthy that LRP1 is also a receptor for APOE, Aβ and is responsible for Tau spreading, although this appears to be through neuronal-mediated endocytosis[134-136]. Engagement of specific Fcγ receptor subtypes is dependent on the IgG subtype, with the degree of activation and subsequent activation further impacted by IgG avidity. Both subunits of the CR3 receptor (Mac-1; ITGAM/ITGB2; CD11b/CD18) have previously been found to have highly correlated and elevated expression in AD[137,138]. Following receptor engagement, Rho-dependent (CR3) or Rac/cdc42-dependent (Fcγ/CR1) signalling in microglia are initiated, leading to clearance of tagged cells/proteins by phagocytosis (ADCP, antibody-dependent cell phagocytosis or CDCP, complement-dependent cell phagocytosis) and/or toxin producing pathways in microglia leading to cell death and phagocytic clearance (ADCC, antibody-dependent cell cytotoxicity or CDCC, complement-dependent cell cytotoxicity). ADCC and CDCC are considered pro-inflammatory and therefore usually only activated to kill pathogens or cells infected by pathogens. Thus, there are many checks and balances to control complement ADCC or CDCC activation because of the particular risk they pose in causing significant bystander damage to tissue and cells[139].
Lipid signals in AD exposed by apoptotic cells (dying neurons), demyelination or metabolic changes are a further source of signalling for microglia during AD pathogenesis. Lipids are a considerably diverse and complex class of molecules having significant structural, metabolic and signalling properties. Phosphatidylserine, phosphatidylethanolamine and sphingomyelin are major components of cell membranes and myelin, a specialist membranous extension of oligodendrocytes[140,141]. Stressed or dying cells, including neurons in AD, can cause “eat me” lipid signals such as phosphatidylserine or phosphatidylethanolamine to become exposed on the cell surface, which mark them out for clearance[142,143]. This can occur following interaction with TREM2[58,144,145] and/or TAM receptors (Tyro3, Axl, and Mertk)[146] and/or brain-specific angiogenesis inhibitor 1 (BAI1) or vitronectin receptor (VNR)[147] on microglia. TREM2 can additionally bind phosphatidic acid, phosphatidylglycerol, phosphatidylinositol, sulfatides[58] and cardiolipin, a component of the outer mitochondrial membrane found in pathogen cell membranes[144]. Furthermore, once phagocytosed, TREM2 knock-out or ageing can cause a failure of lipids such as free cholesterol, cholesteryl esters, and myelin-derived ceramides to be adequately degraded through the lysosomal pathway in microglia. This leads to their accumulation and inflammatory activation[130,148,149].
Response of microglia to pathogens in Alzheimer’s disease
Microglia have a multitude of receptors to detect the wide array of molecules found uniquely on pathogens such as fungi, bacteria, viruses and protozoa. Plasma membrane receptors able to detect pathogens include toll-like receptors (TLRs 1-6), C-type lectin receptors, CLRs (dectin 1, dectin 2), DC-specific ICAM3 grabbing non-integrin (DC-SIGN), mincle and the mannose receptor galectin family proteins (such as galectin 3), CR3, chitin receptors and the class B scavenger receptor CD36. Internal membrane receptors to detect viruses gaining entry include toll-like receptors (TLRs 7-9), RIG-I and MDA5[150,151]. Once engaged by pathogen-specific ligands such as lipopolysaccharide in bacterial cells walls, microglia generate an immediate innate immune response to protect the brain. Furthermore, APP processing leading to amyloid deposition is considered by some to be at the heart of an antimicrobial immune defence mechanism in the brain which can sequester certain microbes in an attempt to neutralise their pathogenicity[152,153]. This has led to the “Antimicrobial Protection Hypothesis of AD” which proposes that viral infectivity lies at the heart of AD aetiology with the deposition of amyloid heralding that infectivity has occurred in an individual[152-155]. In this model, amyloid is a protective mechanism for pathogen entrapment, and together with microglia encasement prevents further infectivity as well as Aβ toxicity, thereby avoiding the need for a much larger and potentially damaging pro-inflammatory response[75-77]. Interestingly, the failure to mount a successful Fc-mediated response in the presence of a viral pathogen such as Herpes Simplex Virus type 1, has recently been suggested as a possible mechanism leading to AD[156]. There is also evidence for AD-associated infectivity by certain viruses. Herpes virus re-activation has greater prevalence in people who develop AD[157-159], infectivity by different strains of herpesvirus causes pathologies reminiscent of AD[153,160,161] and viruses such as West Nile virus have an impact on cognition through complement- and microglia-mediated changes to synapses[47].
Microglia Heterogeneity
Technical advances enabling sequencing of the full repertoire of expression in individual cells has paved the way for defining the molecular identity of immune cells in the brain in health and disease with unprecedented depth and breadth. These data provide important knowledge about cell identity and function, as well as a fantastic resource for the research community. In many cases, authors have publicly provided data for individual gene interrogation via highly accessible Shiny apps, e.g., http://celltypes.org/brain/[162] and http://research-pub.gene.com/BrainMyeloidLandscape[101]. These data provide: (1) a set of exclusive or highly enriched genes for each cell type and subtype; (2) knowledge of genes expressed under different conditions; and (3) the full extent of cell heterogeneity across different microniches, microenvironments and temporal patterns throughout the brain. Knowledge of cell type subsets can be exploited to offer much greater precision to target particular cells whilst sparing others. Whole-genome expression profiling of microglia from normal and disease brain samples has been extensively investigated[163]. This includes non-neurologically affected human brains[71,73,74,164-172], normal mouse brain[71-73,173-182], human brain affected by AD[71,73,74,138,171] or Multiple Sclerosis[164] and AD or other neurodegenerative disease mouse models[71-73,98,100,183], EAE/injury mouse models of Multiple Sclerosis[175,180] or acute injury[182]. Despite using different approaches such as whole sample sequencing with post hoc cell type inference, or prior whole cell or nuclei separation by immunopanning or droplet single cell sorting, many of these studies showed remarkable concordance in the gene expression profiles obtained for different cell types and cell subsets. What emerges from the above human brain studies is stratification of between one and nine major microglia subgroups ranging from homeostatic to activated cell profiles. There is a good deal of conservation in the DAM profile across different disease contexts including developmentally active microglia and in the presence of different pathologies such as in AD (as discussed above). The DAM profile will be further explored below, with respect to AD risk genes.
Genetic Risk of Alzheimer’s Disease
Although largely sporadic, AD has a significant heritable component[184-187]. In rare AD cases, usually with disease onset before 65 years, there are fully penetrant missense or splicing mutations in APP or genes forming part of the γ-secretase complex which cleaves APP (PSEN1 or PSEN2) to a more amyloidogenic fragment (Aβ1-42). For late onset disease (> 65 years), variants in a much larger number of genes has been found, but their contribution to disease risk is generally significantly lower. Over the years, the list of true positive AD-associated risk genes has been extended, consolidated and refined for sporadic AD. In a large part, the risk gene list has evolved following independent validation, improved analytical approaches and a vast expansion in the number of study participants, which minimises false positives and identifies increasingly smaller effect size variants. APOE has been convincingly associated with AD for nearly 30 years[188], reflecting its very large contribution to AD risk. The double SNP haplotype which gives rise to APOEε2, 3 and 4 genotypes confers a 12-fold higher AD risk in those with two APOEε4 alleles, while APOEε2 carriers are protected. Two large studies published around the same time in 2013 identified many gene associations which have continued to be found in subsequent studies[189,190]. Prior to this, ~700 genes had been variously published as having AD association[191]. While some associations were missed by the methods available at the time, many others could not be replicated in these earlier studies. Although substantial for their time, many studies suffered from being statistically underpowered, with small sample sizes leading to stochastic associations. Since then, well-powered and more technically sophisticated gene wide association studies (GWAS) have further consolidated and refined the gene list. They are frequently the product of significantly larger meta-analyses of former cohorts and/or use imputation to improve genome coverage[192-194]. The largest of the recent studies has involved ~500,000 samples from UK Biobank (using parental AD status as a proxy for diagnosis)[195,196]. Whilst GWAS is good at assessing genes with frequency > 5%, more recent AD association studies have used sequencing strategies to identify much rarer high impact variants in genes such as TREM2, PLCG2 (rare protective allele), ABI3 and IL34[193,197-200]. Further work has continued to use family-based approaches which in addition to the well-established mutations in APP, PSEN1 and PSEN2, has begun to expand this list in familial late onset AD[201].
Review of genes associated with Alzheimer’s Disease
We have reviewed GWAS, exome and whole genome sequencing studies and present a list of 84 genes which have been reliably associated with AD in independent and well-powered studies [Table 1]. Studies are from literature searches conducted up to 1st August 2020 in Pubmed, Biorxiv and Medrxiv using the terms “Alzheimer*”, “genetic”, “exome”, “sequencing”, “genome wide association” and “gene”. Only large-scale, familial or genome-wide studies were included. Candidate gene studies were excluded. Appendices and supplementary data from each paper were accessed and included in the analysis. Where associations are SNP-based, SNP-to-gene mapping was performed using the L2G (Locus-to-Gene) score in Open Targets Genetics (OTG) from an index GWAS study if the locus was included[195] or from other papers where the same locus was identified[190,192,194,196]. Genes within each locus were prioritised using the V2G (Variant-to-Gene) score where the L2G (Locus-to-Gene) score was not available from OTG. Prioritisation also considered the SNP-to-gene information from the relevant studies[192,202-204] and the MAGMA (Multi-marker Analysis of GenoMic Annotation) and FUMA (Functional Mapping and Annotation) scores[195].
Review of genes associated with Alzheimer’s Disease
| Locus | Study type | Diagnosis | Lead variant | Closest gene | Variant type | Functional variant suppport? | Most plausible gene | Microglial score | Figure 1 localisation/pathway in microglia | Alternative genes | Microglial score2 | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1q23.2 | GWAS | AD proxy | rs4575098 | ADAMTS4 | 3 Prime UTR Variant | ADAMTS4 | 0 | Not included | DEDD | 0 | [2] | |
| 1q32.2 | GWAS | AD | rs2093760 | CR1 | Intron variant | CR1; PMID: 31104630 PMID: 29675612 | CR1 | 0 | Microglia receptor | CR1L, CD55, YOD1 | 0 | [1,2,3] |
| 1q42.12 | GWGAS | AD | [gene-wise] | ENAH | ENAH | 0 | Not included | [2] | ||||
| 2p22.2 | GWAS | AD + AD proxy | rs876461 | PRKD3 | Intron variant | CEBPZOS | 0 | Not included | PRKD3, NDUFAF7 | 0 | [10] | |
| 2p23.3 | WGS | AD | rs142787485 | RAB10 | Intron variant | RAB10 | 2 | Phagosome | DRC1 | 0 | [46] | |
| 2q14.3 | GWAS | AD | rs4663105 | AC110926.2 | Intergenic | BIN1: PMID: 26101835 | BIN1 | 3 | Phagosome? | [1-5] | ||
| 2q33.1 | WES (familial) | AD | [gene-wise] | CASP8 | Missense variant | CASP8; PMID: 28985224 | CASP8 | 5 | Not included | [38] | ||
| 2q33.2 | GWGAS | AD | [gene-wise] | WDR12 | WDR12 | 0 | Not included | [2] | ||||
| 2q37.1 | GWAS | AD | rs10933431 | INPP5D | Intron variant | INPP5D | 6 | Signalling intermediate | [2-5,13] | |||
| 3p14.3 | GWAS | AD proxy | rs184384746 | HESX1 | Intergenic | IL17RD | 0 | Not included | [2] | |||
| 3q25.31 | GWGAS | AD | [gene-wise] | PLCH1 | PLCH1 | 0 | Not included | [2] | ||||
| 4p15.1 | GWGAS | AD | [gene-wise] | PPARGC1A | PPARGC1A | 0 | Not included | [11] | ||||
| 4p16.1 | GWAS | AD + AD proxy | rs6448453 | CLNK | Intergenic | CLNK | 0 | Not included | HS3ST1 | 0 | [2,10,45] | |
| 4q22.3 | Family-based + GWAS | AD | rs137875858 | UNC5C | Missense variant | UNC5C; PMID: 25419706 | UNC5C | 0 | Not included | [41] | ||
| 4q24 | WES (familial) | AD | [gene wise] | TACR3 | Missense variant | TACR3; 10.1101/2020.01.28.923789 | TACR3 | 0 | Not included | [30] | ||
| 4q24 | WES (familial) | AD+ NDother | [gene wise] | TET2 | Missense variant | TET2; PMID: 32330418 | TET2 | 0 | Not included | [33] | ||
| 5q13.1 | GWAS | AD | rs4704171 | ANKRD31 | Intron variant | ANKRD31 | 0 | Not included | GCNT4 | 0 | [1] | |
| 5q14.3 | GWAS | AD | rs190982 | MEF2C-AS1 | Intron variant | MEF2C | 2 | Transcription factor | [5,13] | |||
| 5q31.3 | GWAS | AD | rs11168036 | HBEGF | Intergenic | HBEGF | 0 | Not included | PFDN1 | 0 | [6,44] | |
| 6q14.1 | GWGAS | AD | [gene-wise] | TPBG | TPBG | 0 | Not included | [6] | ||||
| 6p22.3 | GWGAS | AD | [gene-wise] | PRL | PRL | 0 | Not included | [2] | ||||
| 6p21.32 | GWAS | AD | rs6931277 | HLA-DQA1 | Intergenic | HLA-DQA1 | 5 | Microglia receptor | HLA-DRB5, HLA-DRA, HLA-DRB1 | 5 | [2-5] | |
| 6p21.1 | WES | AD | [gene-wise] | TREML2 | Missense variant | PMID: 24439484 | TREML2 | 5 | Microglia receptor | [20] | ||
| 6p21.1 | GWAS | AD+AD proxy | rs187370608 | LOC101929555 | intron Variant | TREM2; PMID: 23150908 PMID: 23150934 | TREM2 | 6 | Microglia receptor | APOBEC2, TREML2 | 5 | [2,3] |
| 6p21.1 | GWAS | AD | rs75932628 | TREM2 | missense Variant | TREM2; PMID: 23150908 PMID: 23150934 | TREM2 | 6 | Microglia receptor | [7,9,21] | ||
| 6p12.3 | GWAS | AD | rs9381563 | CD2AP | intergenic | CD2AP: PMID: 26101835 | CD2AP | 0 | Not included | [1-3,31] | ||
| 7p22.1 | GWGAS | AD | [gene-wise] | WIPI2 | WIPI2 | 0 | Not included | [2] | ||||
| 7p22.3 | GWGAS | AD | [gene-wise] | TMEM184A | TMEM184A | 0 | Not included | [2] | ||||
| 7p14.1 | GWAS | AD | rs2718058 | NME8 | Intergenic | GPR141 | 5 | Microglia receptor? | [5] | |||
| 7p21.3 | GWAS+ | LOAD subtype | rs1990620 | TMEM106B | Intron variant | TMEM106B | 0 | Not included | [41] | |||
| 7q11.23 | GWGAS | AD | [gene-wise] | RHBDD2 | Intron variant | RHBDD2 | 0 | Not included | [36] | |||
| 7q22.1 | GWGAS | AD | [gene-wise] | ZNF655 | ZNF652 | 0 | Not included | [9] | ||||
| 7q22.1 | GWAS | AD | rs1859788 | PILRA | Missense variant | PILRA; PMID: 29181857 PMID: 30388101 | PILRA | 6 | Microglia receptor | [2-5,13] | ||
| 7q35 | GWAS | AD | rs7810606 | EPHA-AS1 | Intron variant | EPHA1: PMID: 26101835 | EPHA1 | 0 | Not included | FAM131B, ZYX | 0 | [1-5,8] |
| 7q35 | GWAS | AD+AD proxy | rs114360492 | CNTNAP2 | Intron variant | CNTNAP2 | 0 | Not included | [1,2] | |||
| 7q36.3 | WES | AD | 7:154988675:G:A | AC099552.4 | AC099552.4 | 0 | Not included | [9] | ||||
| 8p21.1 | GWAS | AD | rs28834970 | PTK2B | Intron variant | PTK2B | 0 | Signalling intermediate | TRIM35 | 0 | [1,5,6] | |
| 8p21.2 | GWAS | AD | rs4236673 | CLU | Intron variant | CLU | 0 | Not included | [1-5,7] | |||
| 8q22.1 | GWAS | AD | rs10098778 | NDUFAF6 | Intron variant | TP53INP1 | 0 | Not included | NDUFAF6 | 0 | [1] | |
| 8q24.3 | GWAS | AD+AD proxy | rs34173062 | SHARPIN | Missense variant | SHARPIN; PMID: 31216982 https://doi.org/10.1101/196410 | SHARPIN | 0 | Not included | [10,35,42] | ||
| 10p14 | GWAS | AD | rs11257238 | USP6NL | Intergenic | USP6NL | 0 | Not included | ECHDC3 | 0 | [6,3,44] | |
| 10q21.2 | GWGAS | [gene-wise] | CCDC6 | CCDC6 | 0 | Not included | [2] | |||||
| 10q22.1 | GWGAS | AD | [gene-wise] | CDH23 | CDH23 | 0 | Not included | [36] | ||||
| 10q25.3 | WES (familial) | AD | [gene wise] | CASP7 | Missense variant | CASP7; PMID: 30503768 | CASP7 | 0 | Not included | [16] | ||
| 11p11.2 | GWAS | AD | rs12292911 | PSMC3 | Intergenic | SPI1: doi: 10.1038/nn.4587 | SLC39A13 | 0 | Not included | SPI1 | 6 | [3-5] |
| 11q12.2 | GWAS | AD | rs2081545 | MS4A6A | Intergenic | MS4A4A | 5 | Microglia receptor | [1-6,8] | |||
| 11q14.2 | GWAS | AD | rs867611 | PICALM | Intron variant | PICALM | 2 | Not included | EED | 0 | [1-6,8,31] | |
| 11q24.1 | GWAS | AD | rs11218343 | SORL1 | Intron variant | SORL1; PMID: 22472873 PMID: 28595629 | SORL1 | 3 | Phagosome Receptor | [1-5] | ||
| 14q11.2 | GWGAS | AD | [gene wise] | C14orf93 | C14orf93 | 0 | Not included | [2] | ||||
| 14q22.1 | GWAS | AD | rs17125924 | FERMT2 | intron variant | FERMT2 | 0 | Not included | STYX | 0 | [3-5,13] | |
| 14q24.1 | GWGAS | AD | [gene-wise] | PSEN1 | PSEN1; PMID: 32020597 | PSEN1 | 0 | Not included | [34] | |||
| 14q32.12 | GWAS | AD | rs12590654 | SLC24A4 | intron variant | RIN3; PMID: 28738127 | RIN3 | 6 | Phagosome? | SLC24A4 | 0 | [2-5] |
| 14q32.33 | GWGAS | AD | [gene-wise] | IGHV1-67 | IGHV1-67 | 0 | Not included | [12] | ||||
| 14q32.33 | WES | AD | rs12890621 | IGHG3 | missense variant | IGHG3: PMID: 30108311 | IGHG3 | 0 | Not included | [9] | ||
| 15q21.2 | GWAS | AD proxy | rs10467994 | SPPL2A | intron variant | SPPL2A | 0 | Not included | [2,3,44] | |||
| 15q21.3 | GWAS | AD | rs442495 | ADAM10 | intron variant | ADAM10 | 0 | Microglia receptor Cleavage | [2-4] | |||
| 15q22.2 | GWAS | AD proxy | rs117618017 | APH1B | missense variant | APH1B: PMID: 30617256 | APH1B | 0 | Not included | RAB8B | 0 | [2] |
| 15q22.2 | GWGAS | AD | [gene-wise] | RORA | RORA | 0 | Not included | [11] | ||||
| 15q22.31 | GWAS | AD | rs74615166 | TRIP4 | intron variant | CSNK1G1 | 0 | Not included | TRIP4 | 0 | [14] | |
| 16p12.3 | GWAS | AD | rs7185636 | IQCK | intron variant | IQCK | 0 | Not included | [4] | |||
| 16p11.2 | GWAS | AD proxy | rs59735493 | KAT8 | intron variant | KAT8 | 0 | Not included | FBXL19, BCL7C, TAOK2, ZNF646 | 0 | [2,3] | |
| 16q12.1 | GWGAS | AD | [gene-wise] | ZNF423 | ZNF423 | 0 | Not included | [11] | ||||
| 16q22.1 | GWAS | AD+AD proxy | rs4985556 | IL34 | missense variant | IL34; PMID: 29777097 https://doi.org/10.1101/19012021 | IL34 | 0 | Microglia receptor Ligand | [2,3,10] | ||
| 16q23.1 | GWAS | AD | rs62039712 | WWOX | intergenic | MAF | 6 | Transcription factor | WWOX | 0 | [4] | |
| 16q23.3 | GWAS | AD | rs72824905 | PLCG2 | missense variant | PLCG2; PMID: 28714976 | PLCG2 | 6 | Cytoskeletal regulator | [7] | ||
| 17p13.2 | GWAS | AD+AD proxy | rs72835061 | CHRNE | intron variant | PLD2 | 0 | Not included | CHRNE, ZFP3, ZNF653, USP6, ZNF652, ZNF232 | 0 | [2,3] | |
| 17p13.2 | GWAS | AD+AD proxy | rs113260531 | LOC100130950 | intron variant | RABEP1 | 0 | Not included | SCIMP | 5 | [1,10] | |
| 17q21.31 | GWGAS | AD | [gene-wise] | SPPL2C | SPPL2C | 0 | Not included | [30] | ||||
| 17q21.31 | WES-Non-APOE*4 | AD | rs199533 | NSF | missense variant | NSF | NSF | 0 | Not included | [37] | ||
| 17q21.32-33 | GWAS | AD | rs616338 | ABI3 | missense variant | ABI3; PMID: 28714976 | ABI3 | 6 | Cytoskeletal regulator | [7] | ||
| 17q21.32-33 | GWAS | AD | rs28394864 | AC091180.5 | intron variant | ZNF652 | 0 | Not included | [2] | |||
| 17q22 | GWAS | AD | rs2632516 | TSPOAP-AS1 | intron variant | BZRAP1 | 0 | Not included | MPO, SUPT4H1, TRIM37, TSPOAP1 | 0 | [2,6] | |
| 17q23.2 | WES | AD | [gene-wise] | ERN1 | ERN1 | ERN1 | 0 | Not included | [30] | |||
| 17q23.3 | GWAS | AD | rs6504163 | ACE | Intergenic | ACE | 0 | Not included | PSMC5, CYB561 | 0 | [3,4] | |
| 17q25.1 | WGS | AD | [gene-wise] | SLC9A3R1 | intron Variant | SLC9A3R1 | 0 | Not included | [36] | |||
| 18q12.1 | GWAS | AD | rs8093731 | DSG2 | intron Variant | DSG2 | 0 | Not included | [2,4] | |||
| 18q21.31-21.32 | GWAS | AD proxy | rs76726049 | ALPK2 | intron Variant | ALPK2 | 0 | Not included | [2] | |||
| 19p11.3 | GWAS | AD | rs111278892 | CNN2 | intergenic | ABCA7: PMID: 26101835 PMID: 25807283 PMID: 27289440 | ABCA7 | 0 | Not included | CNN2 | 0 | [1,2] |
| 19p13.3 | GWAS - APOE4 int. | AD | rs9749589 | NFIC | Intron variant | NFIC | 0 | Not included | [6] | |||
| 19p13.3 | GWAS | AD | rs5117 | APOC1 | Intron variant | APOE | APOE | 2 | Microglia receptor ligand | [1] | ||
| 19q13.2 | WES (familial) | AD | PLD3 | PLD3; PMID: 24336208 | PLD3 | 0 | Not included | [40] | ||||
| 19q13.41 | GWAS | AD | rs3865444 | CD33 | Intergenic | CD33; PMID: 28747436 PMID: 24381305 | CD33 | 5 | Microglia receptor | [1-5,8] | ||
| 20q11.2-q13.31 | GWAS | AD | rs6014724 | CASS4 | Intron variant | CASS4 | 5 | Cytoskeletal Regulator | [2-5,31] | |||
| 21q21.3 | GWAS | AD | rs2830500 | ADAMTS1 | Intergenic | ADAMTS1 | 0 | Not included | ADAMTS5 | 0 | [4] | |
| 21q21.3 | GWAS | AD | rs2154481 | APP | Intron variant | APP | APP | 0 | Not included | [10] |
An over-representation of risk genes with lipid and/or immune function was first recognised in 2010[205]. Since then, further consolidation of the list of risk genes and the addition of genes with rare risk variants has further reinforced this finding. In fact, there is even stronger evidence for the immune system and microglia having a role in AD vulnerability in the list we have assembled (details below and in Table 1). Whilst knowledge of these risk variants in these genes may never be useful by themselves for establishing individual clinical diagnosis, they nevertheless have considerable value in refining pathways of vulnerability which can be targeted for therapeutic development, for identifying and enriching high risk participants for clinical trials related to these pathways and supporting diagnosis as part of a panel of factors including clinical and biomarker assessments as part of a composite risk score.
The degree of knowledge about the function of each gene is highly variable. Nevertheless, by combining evidence from the literature, and integrating this with transcriptomic and biochemical data, it may be possible to achieve a cell- and systems-level understanding of how these genes are linked in pathways that are responsible for the brain immune response in AD which could be exploited therapeutically.
Expression of Alzheimer’s Disease Risk Genes in Microglia
With advances in single-cell isolation and sequencing, it is now possible to establish which of the AD risk genes have unique or highly enriched expression in microglia in the brain. To achieve this, we used published data from FACS-sorted cell populations isolated from human donor parietal cortex[172,206]. Genes were given a score reflecting whether they met the criteria used by the authors for microglial expression[172], our own internal criteria of RPKM (Reads Per Kilobase of transcript, per Million mapped reads) (either RPKM > 10 or > 20 RPKM) or a 4-8- 10- or 20-fold-increase in differential expression (DE) over background. Genes meeting any of these criteria are shown in bold, along with their microglia score using these criteria (0-6) [Table 1]. Twenty (shown in bold throughout this review) out of the eighty-four genes linked to AD risk variants had a microglia enrichment score ≥ 1 [Table 1]. In separate data generated by us, we confirm the most highly expressed of these genes with a score of 6 (INPP5D, TREM2, PILRA, RIN3, MAF, PLCG2, ABI3) had highly correlated co-expression with other microglia genes in whole human brain hippocampal tissue, either from neurologically normal or AD donors (unpublished data). Other studies also confirmed these genes have high or enriched expression in microglia from normal brain tissue[166,167]. Some of them also appear to have highly correlated and enriched expression with genes more likely to be found in particular microglia subsets in normal brain - either in highly abundant homeostatic microglia (Cluster 3: BIN1, INPP5D, MEF2C, SORL1, MAF, PLCG2), age-associated homeostatic cells with high levels of MHC Class II expression (Cluster 6: RAB10, TREM2, RIN3, APOE, CD33) or in a small population of pro-inflammatory microglia (Cluster 8: CASS4, CASP8, PILRA)[165] suggesting there may be multiple routes in microglia contributing to greater AD vulnerability.
Functional Impact of Risk Variants in Microglia Genes
For some genes and risk variants, the functional consequence of the risk variant(s) and knowledge about the role of the protein in normal and AD brain is quite well established (TREM2 and CD33, discussed below). While for other genes where the associated “tag” SNP is intergenic (BIN1, HLA-DQA1, GPR141, MS4A4A, MAF), the causal driver of the association is far more opaque. In many cases, considerable experimental work is still needed to link the tag SNP to the causal association. Furthermore, it is important to emphasise that as more information comes to light, the gene responsible for the association may also change as the causal variant becomes known. For coding SNPs, particularly missense variants (CASP8, TREML2, TREM2, PILRA, PLCG2, ABI3) or where a stop codon leads to a truncated protein (IL34), the tag and causal SNP are likely to be one and the same. In silico evaluation of the functional consequences of amino acid substitution (using software such as SIFT, PolyPhen, REVEL, MetaLR, MutationAssessor and PROVEAN linked with output integrated within NCBI Ensembl)[207-212] can be a good first step to providing evidence for pathogenicity of any SNP identified by association analysis.
In TREM2, a number of rare hypomorphic AD missense risk variants have been shown to impact TREM2 production (rs104894002, Q33X), expression or turnover at the cell surface (rs75932628, R47H)[115,213], α-secretase cleavage of the extracellular soluble ectodomain (rs2234255, H157Y)[115,214,215] and/or ligand binding (rs75932628, R47H; rs143332484, R62H; rs2234253/rs2234258/rs2234256, T96K/W191X/L211P; rs2234255, H157Y)[216-218]. Additional rare missense variants affecting both alleles primarily alter protein structure, cell surface expression and shedding, and cause Nasu-Hakola disease[115,216,219]. In CD33, the intergenic tag SNP (rs3865444) is in perfect linkage disequilibrium with an intronic SNP (rs12459419) which affects splicing of exon 2[220]. The risk allele leads to inclusion of exon 2 which codes for an extracellular Ig V-set domain and a longer isoform (designated CD33M) which has higher cell surface expression. The protective allele missing from this domain due to the exclusion of exon 2 leads to a shorter isoform (designated CD33m). The Ig V-set domain is important for the sialic acid-binding properties of CD33, but may also be important for changing the conformation of ligand binding to CD33 and thereby a shift between inhibitory and activation signalling in microglia which has important consequences for AD risk[221].
Pathways Connecting Alzheimer’s Disease Risk Genes in Microglia
It is possible to link a number of the microglia specific/enriched genes within key microglia signalling pathway(s), either from direct evidence or through their homology with proteins where greater functional data exists (e.g., ABI3 cellular localisation and function can be inferred from family members ABI1 and ABI2) [Figure 1 and Table 1]. This is a starting point for gaining a holistic understanding of how microglia behaviour may impact brain function and the disease process. It will also support the development of testable hypotheses including possible treatment targets. The pathways will be refined further in the coming years as greater functional knowledge of each gene and pathways emerges. Many of the microglia enriched/specific AD risk genes are linked to PAMP/DAMP responsive cell membrane receptors linked to phagocytosis and the cell processes necessary for phagocytosis - cytoskeletal machinery, endocytosis and lysosomal degradation within specialist phagosomes as well as changes in transcription and secretory pathways. In this way, we are beginning to consolidate knowledge about microglia that present opportunities to manipulate them towards a more protective response in AD. For example, TREM2 activating antibodies[222,223] including the “first in man” Phase I clinical study recently published for agonistic monoclonal antibody, AL002c[119].
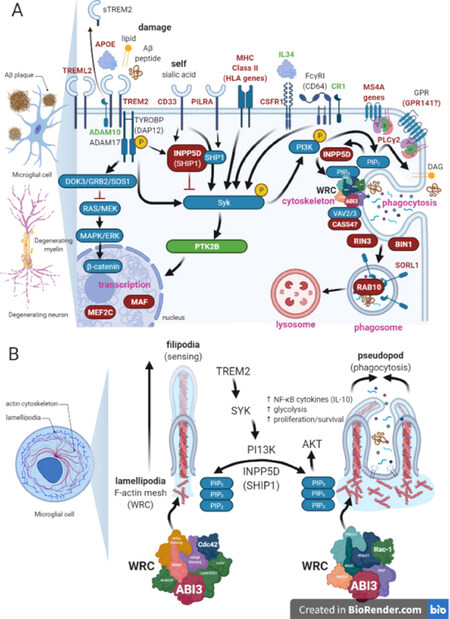
Figure 1. Microglia signalling pathways involving AD risk genes. (A) Key signalling pathways in microglia involving AD risk genes (microglia enriched highlighted in blue, other AD risk genes, purple); (B) signalling converging on the Wave Regulatory Complex (WRC) can impact cytoskeletal polymerisation/depolymerisation to inhibit lamellipodia capping and promote formation of filopodia for cell sensing (homeostatic microglia) or promote lamellipodia capping which promotes phagocytosis in response to damage/pathogen signals (activated microglia).
The majority of the microglia risk genes code for receptors or proteins which associate with receptors at the inner cell membrane, having transited through internal membranes such as ER, Golgi or endosomes before becoming active on the cell surface (TREM2, TREML2, CD33, MS4A4A, INPP5D, HLA-DQA1, GPR141, PILRA, PICALM, SORL1, PLCG2) [Figure 1A]. Frequently, the ectodomains of these receptors are shed by proteolytic processing by secretases at the cell surface (e.g. ADAM10 cleaves TREM2 while SORL1 is cleaved by presenilin activity)[115,214,215,224-226]. The functional significance of this is not yet fully understood. Some ligands for these receptors including PAMPs and DAMPs are known. For example, TREM2 is activated by damage-associated anionic and zwitterionic lipids, including sphingomyelin, phosphatidylserine and phosphatidylethanolamine, which are likely to be displayed by damaged myelin or apoptotic cells such as dying neurons[58,59,144,145]. Interestingly, in addition to being a receptor for sialic-acid containing proteins such COLEC12, NPDC1, CLEC4G, CD99, complement component 4A and PIANP, PILRA also appears to be a co-receptor for the cell entry of Herpes virus[227] and thus could be a potential initiator of AD in the viral-induced model of pathogenesis (discussed above). The AD missense risk variant (rs1859788; G78R) results in reduced ligand binding to PILRA, although it isn’t yet clear how this would impact AD risk[228].
Many of these receptors share the same adaptor proteins and/or downstream signalling cascades and they appear to propagate complex inhibitory or activating signals depending on the avidity of ligand binding, as reported for TREM2 and CD33[221]. TYROBP (DAP12) is in fact a common adaptor for many immune cell receptors which do not have their own ITAM domain and therefore potentially adds further biological complexity as it presumably must respond and coordinate the competing needs of different receptors simultaneously. It is also common for immune receptors (including those coded for by AD microglia risk genes) to signal via the phosphatidylinositol signalling pathway. The phosphatidylinositols are important lipid sensing messengers, regulating many cell processes reliant on cytoskeletal dynamics - sensing, phagocytosis, cell division, vesicle trafficking, adhesion and motility[229,230]. The key elements of this pathway are the conversion of PIP2 (phosphatidylinositol 4,5-bisphosphate) to second messengers IP3 (inositol 1,4,5-trisphosphate) and DAG (diacylglycerol) by PLC enzymes, of which the AD risk gene PLCG2 is a member, or, conversion of PIP2 to PIP3 [Phosphatidylinositol (3,4,5)-trisphosphate] by the enzyme PI3K [Figure 1A].
TREM2-DAP12 signalling triggers many protein and lipid phosphorylation cascades causing calcium mobilization, integrin activation, mTOR and MAPK signalling, activation of energetic metabolism[107] and cytoskeleton rearrangement via Syk[109] and is thus considered necessary for protection against damage and pathology in AD. PI3K activation via Syk is considered an important downstream target of TREM2[231,232], expected to convert PIP2 to PIP3 and subsequently the type of membrane extensions from lamellipodia.
Lamellipodia are structures at the inner membrane leaflet. They consist of a mesh of F-actin and interacting proteins, which give membranes their irregular appearance at high magnification. It is from here that processes/filopodia can develop when F-actin capping is blocked, allowing actin polymerisation to continue. The balance between lamellipodia and process/filopodia formation is regulated by the large multiprotein Wave Regulatory Complex (WRC) [Figure 1A]. Microglia processes/filopodia are dynamic membrane protrusions extending (actin polymerisation) and retracting (actin depolymerisation) 100mm/3mm from the cell soma or process end, respectively[233]. Processes/filopodia are characteristic of homeostatic/ramified microglia, allowing microglia to efficiently survey their territories for signals. Their formation relies on high local levels of PIP2 at the inner cell membrane leaflet. PIP2 to PIP3 conversion generates shorter extensions or podosomes to develop, which allow the membrane to collapse to form phagocytic cups able to engulf and internalise damaged material or pathogens [Figure 1B]. The composition and activity of the WRC can change according to the signals received, either favouring the clustering of PIP2 or PIP3. Signalling through cdc42 via WRC: cdc42-mDia2 (formin)-WASP (WAS)-N-WASP (Eps8/SOS1, VASP, ABI3) clusters PIP2 and favours filopodia formation, while signalling through Rac via WRC: Rac-IRSp53-WAVE-Arp2/3-ABI3-NAP-PIR121) clusters PIP3 and favours lamellipodia[234-236]. Loss of TREM2 reduces the degree of membrane ruffling, leads to increased filopodia number with reduced length and impairs phagocytic capability when BV2 microglia are stimulated with ATP or M-CSF (CSF1)[237]. Likewise, an absence of TREM2 impairs formation of actin rings and podosomes in osteoclasts, essential for bone resorption[238], consistent with a block in PIP2 to PIP3 which appears to be essential for “sealing off” the membrane edges.
PI3K activity can be inhibited by the AD risk gene INPP5D (SHIP-1), which can cooperate with the protein SHP-1 to inhibit immune receptor signalling. In fact, SHP-1 is a key downstream effector of CD33, at least when activated through the Ig V-set domain (discussed above). PI3K activity is complex, but as indicated above, downstream signalling is expected to regulate PIP2 to PIP3, as well as regulating other complexes which promote membrane flexibility. SHIP-1 is also a phosphatase involved in PIP2 to PIP3 production. Loss of SHIP-1 activity might therefore be expected to lead to a failure in PIP3 production leading to a block in phagocytosis.
ABI3, another microglia specific AD risk gene, appears to physically interact with VASP within the WRC which controls cytoskeletal activity[239-241][Figure 1]. ABI3 activity is expected to favour Cdc42-dependent filopodia formation since high levels of ABI3 decrease phosphorylation of Src, Lyn, Lyk, Yes, Hck, Fyn, Pyk2, FAK and FGR and members of P13K/AKT, AMPK, p38 MAPK, SAPK/JNK, mTOR, consistent with a role in maintaining high levels of PIP2 permissive for filopodia formation in homeostatic microglia[242]. When ABI3 is phosphorylated at residue p.S342 by AKT, it has reduced association with the WRC[242]. This would be expected to be permissive for PIP3 and impact not only phagocytosis and cell morphology, but also potentially cell motility and cytokinesis[239]. Although it still needs to be tested for ABI3, it is possible that the phosphorylation of ABI3 may also be modulated by TREM2-dependent activity, but this and the functional impact of the missense AD risk variant rs616338; c.626C>T; p.S209F which may abolish such signalling needs further testing.
Human Microglia Cell Models
In many ways, we have entered a new golden age for microglia research. Not only have they become centre stage for their clear contributions to disease and the remarkable new insights in to their roles in modifying human behaviours[41], but new knowledge about their developmental origin and expression profiles have paved the way for authentic human microglia to be generated from blood or other accessible tissue samples[243-249]. Microglia are myeloid cells formed in the yolk sac during the first wave of haematopoiesis[17]. They share close molecular identity with other haematopoietic cells and specialist macrophages in different tissue niches, such as osteoclasts in bone and Kupffer cells in liver. However, different macrophages can exhibit a unique response, such as found with albumin exposure[250]. Human iPSC-microglia are becoming a well-established tool to understand the molecular events linking microglia activation with signalling pathways and functional outcomes. They appear to display many of the expected morphological and functional behaviours and express genes expected of authentic human microglia, where comparisons have been made with primary ex vivo foetal and/or adult microglia or isolated and profiled donor post mortem cells[243,245-247,249]. They can also be grown in co-culture which allows more complex investigations in to their relationship with other brain cell types[251]. Importantly, they can be generated from patient samples, including those with different genetic risk variants So overall, they offer a step change in the breadth of research questions which can be investigated and increased confidence in the results which accurately capture human microglia activities.
Conclusion
Genetic findings provide very clear evidence that fundamental processes controlling how microglia behave can impact vulnerability to AD. How microglia recognise and respond to age-associated signals determines whether they are ultimately protective or damaging in the brain and provide hope that microglia behaviour is modifiable. Identifying the functional links between AD risk genes, their causal variants and the pathways they operate in, particularly those highly expressed by microglia, will pave the way for identifying tractable targets and new approaches for therapeutic development. Methodological advances in generating authentic human microglia and the ability to selectively survey every gene they express under different conditions supports these efforts.
Declarations
AcknowledgmentsThis work was supported by the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115976, PHAGO. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.
Authors’ contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation and drafted the manuscript: Hodges AK
Made substantial contributions to conception and design of the study and performed data analysis and interpretation and revised the manuscript for intellectual content: Collier D, Pocock JM
Revised the manuscript for intellectual content: Piers TM, Cousins O
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115976, PHAGO. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.
Conflicts of interestDavid Collier is an employee of Eli Lilly and Company. All authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Wendeln AC, Degenhardt K, Kaurani L, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018;556:332-8.
2. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol 2017;18:123-31.
3. van Langelaar J, Rijvers L, Smolders J, van Luijn MM. B and T cells driving multiple sclerosis: identity, mechanisms and potential triggers. Front Immunol 2020;11:760.
4. Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol 2014;10:579-96.
5. Gelb S, Stock AD, Anzi S, Putterman C, Ben-Zvi A. Mechanisms of neuropsychiatric lupus: The relative roles of the blood-cerebrospinal fluid barrier versus blood-brain barrier. J Autoimmun 2018;91:34-44.
6. Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer 2020;20:26-41.
8. Forrester JV, McMenamin PG, Dando SJ. CNS infection and immune privilege. Nat Rev Neurosci 2018;19:655-71.
9. Viggars AP, Wharton SB, Simpson JE, et al. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci Lett 2011;505:25-30.
10. Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med 2017;214:3151-69.
11. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 2011;12:723-38.
12. Yang AC, Stevens MY, Chen MB, et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 2020;583:425-30.
13. Laman JD, Weller RO. Drainage of cells and soluble antigen from the CNS to regional lymph nodes. J Neuroimmune Pharmacol 2013;8:840-56.
14. Greenhalgh AD, David S, Bennett FC. Immune cell regulation of glia during CNS injury and disease. Nat Rev Neurosci 2020;21:139-52.
15. Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015;212:991-9.
16. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci 2017;20:136-44.
17. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841-5.
18. Schulz C, Gomez Perdiguero E, Chorro L, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012;336:86-90.
19. Lawson L, Perry V, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990;39:151-70.
20. Réu P, Khosravi A, Bernard S, et al. The lifespan and turnover of microglia in the human brain. Cell Rep 2017;20:779-84.
21. Askew K, Li K, Olmos-Alonso A, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep 2017;18:391-405.
22. Lawson L, Perry V, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992;48:405-15.
23. Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 2009;87:181-94.
24. Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol 2014;258:5-16.
25. Neumann H, Takahashi K. Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 2007;184:92-9.
26. Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci 2007;30:527-35.
27. Ribeiro M, Brigas HC, Temido-Ferreira M, et al. Meningeal γδ T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci Immunol 2019;4:eaay5199.
28. Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 2016;353:766-71.
29. Derecki NC, Cardani AN, Yang CH, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med 2010;207:1067-80.
30. Eyo UB, Gu N, De S, Dong H, Richardson JR, Wu LJ. Modulation of microglial process convergence toward neuronal dendrites by extracellular calcium. J Neurosci 2015;35:2417-22.
31. Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci 2014;34:10528-40.
32. Li Y, Du XF, Liu CS, Wen ZL, Du JL. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell 2012;23:1189-202.
33. Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 2009;29:3974-80.
34. Tremblay MÈ, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol 2010;8:e1000527.
35. Madry C, Kyrargyri V, Arancibia-Cárcamo IL, et al. Microglial ramification, surveillance, and interleukin-1β release are regulated by the two-pore domain K+ channel THIK-1. Neuron 2018;97:299-312.e6.
36. Akiyoshi R, Wake H, Kato D, et al. Microglia enhance synapse activity to promote local network synchronization. eNeuro 2018;5:ENEURO.
37. Badimon A, Strasburger HJ, Ayata P, et al. Negative feedback control of neuronal activity by microglia. Nature 2020;586:417-23.
38. Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011;333:1456-8.
39. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012;74:691-705.
40. Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018;359:1269-73.
41. Wang C, Yue H, Hu Z, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020;367:688-94.
42. Weinhard L, di Bartolomei G, Bolasco G, et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 2018;9:1228.
43. Nguyen PT, Dorman LC, Pan S, et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 2020;182:388-403.e15.
44. Miyamoto A, Wake H, Ishikawa AW, et al. Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun 2016;7:12540.
45. Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013;155:1596-609.
46. Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007;131:1164-78.
47. Vasek MJ, Garber C, Dorsey D, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016;534:538-43.
48. Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016;352:712-6.
49. Li Q, Cheng Z, Zhou L, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 2019;101:207-223.e10.
50. Nemes-Baran AD, White DR, DeSilva TM. Fractalkine-dependent microglial pruning of viable oligodendrocyte progenitor cells regulates myelination. Cell Rep 2020;32:108047.
51. Hughes AN, Appel B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat Neurosci 2020;23:1055-66.
52. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 2020;20:95-112.
53. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010;10:826-37.
54. Lessard CB, Malnik SL, Zhou Y, et al. High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol Med 2018;10:e9027.
55. Zhao Y, Wu X, Li X, et al. TREM2 is a receptor for β-Amyloid that mediates microglial function. Neuron 2018;97:1023-1031.e7.
56. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 2016;91:328-40.
57. Atagi Y, Liu CC, Painter MM, et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem 2015;290:26043-50.
58. Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015;160:1061-71.
59. Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE. Pattern recognition by TREM-2: binding of anionic ligands. J Immunol 2003;171:594-9.
60. Derk J, MacLean M, Juranek J, Schmidt AM. The receptor for advanced glycation endproducts (RAGE) and mediation of inflammatory neurodegeneration. J Alzheimers Dis Parkinsonism 2018;8:421.
61. Qi XM, Ma JF. The role of amyloid beta clearance in cerebral amyloid angiopathy: more potential therapeutic targets. Transl Neurodegener 2017;6:22.
62. Frautschy SA, Yang F, Irrizarry M, et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 1998;152:307-17.
63. Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH, et al. Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol 1988;132:86-101.
64. Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol 1989;77:569-75.
65. Itagaki S, Mcgeer P, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. Journal of Neuroimmunology 1989;24:173-82.
66. Wegiel J, Wisniewski HM. The complex of microglial cells and amyloid star in three-dimensional reconstruction. Acta Neuropathol 1990;81:116-24.
67. Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 1999;154:1673-84.
68. Bolmont T, Haiss F, Eicke D, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 2008;28:4283-92.
69. Füger P, Hefendehl JK, Veeraraghavalu K, et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci 2017;20:1371-6.
70. Fuhrmann M, Bittner T, Jung CK, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci 2010;13:411-3.
71. Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s Disease. Cell 2017;169:1276-1290.e17.
72. Mathys H, Adaikkan C, Gao F, et al. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep 2017;21:366-80.
73. Zhou Y, Song WM, Andhey PS, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 2020;26:131-42.
74. Grubman A, Chew G, Ouyang JF, et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci 2019;22:2087-97.
75. Wang Y, Ulland TK, Ulrich JD, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 2016;213:667-75.
76. Yuan P, Condello C, Keene CD, et al. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016;90:724-39.
77. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun 2015;6:6176.
78. Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 2013;8:e60921.
79. Daria A, Colombo A, Llovera G, et al. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J 2017;36:583-603.
80. Grathwohl SA, Kälin RE, Bolmont T, et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci 2009;12:1361-3.
81. Caldeira C, Cunha C, Vaz AR, et al. Key Aging-Associated Alterations in Primary Microglia Response to Beta-Amyloid Stimulation. Front Aging Neurosci 2017;9:277.
82. Floden AM, Combs CK. Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J Alzheimers Dis 2011;25:279-93.
83. Venegas C, Kumar S, Franklin BS, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017;552:355-61.
84. Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010;330:1774.
85. Boche D, Nicoll JA. The role of the immune system in clearance of Abeta from the brain. Brain Pathol 2008;18:267-78.
86. Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 2000;6:916-9.
87. Bacskai BJ, Kajdasz ST, Christie RH, et al. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 2001;7:369-72.
88. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 2003;9:448-52.
89. Nicoll JA, Barton E, Boche D, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol 2006;65:1040-8.
90. Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999;400:173-7.
91. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016;537:50-6.
92. Wilcock DM, Rojiani A, Rosenthal A, et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci 2004;24:6144-51.
93. Hopp SC, Lin Y, Oakley D, et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflammation 2018;15:269.
94. Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 2015;18:1584-93.
95. Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015;138:1738-55.
96. Bolós M, Llorens-Martín M, Jurado-Arjona J, Hernández F, Rábano A, Avila J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J Alzheimers Dis 2016;50:77-87.
97. Mathys H, Davila-Velderrain J, Peng Z, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019;570:332-7.
98. Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE Pathway drives the transcriptional phenotype of dysfunctional microglia in Neurodegenerative Diseases. Immunity 2017;47:566-581.e9.
99. Chen WT, Lu A, Craessaerts K, et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s Disease. Cell 2020;182:976-991.e19.
100. Carbajosa G, Malki K, Lawless N, et al. Loss of Trem2 in microglia leads to widespread disruption of cell coexpression networks in mouse brain. Neurobiol Aging 2018;69:151-66.
101. Srinivasan K, Friedman BA, Etxeberria A, et al. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep 2020;31:107843.
102. El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med 2003;197:1657-66.
103. Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 2010;11:155-61.
104. Sheedy FJ, Grebe A, Rayner KJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 2013;14:812-20.
105. Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019;575:669-73.
106. Yan SD, Chen X, Fu J, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996;382:685-91.
107. Ulland TK, Colonna M. TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol 2018;14:667-75.
108. Piers TM, Cosker K, Mallach A, et al. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J 2020;34:2436-50.
109. Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol Cell 2008;31:422-31.
110. Schlepckow K, Monroe KM, Kleinberger G, et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med 2020;12:e11227.
111. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005;201:647-57.
112. Garcia-Reitboeck P, Phillips A, Piers TM, et al. human induced pluripotent stem cell-derived microglia-like cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Rep 2018;24:2300-11.
113. Xiang X, Werner G, Bohrmann B, et al. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med 2016;8:992-1004.
114. Cantoni C, Bollman B, Licastro D, et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol 2015;129:429-47.
115. Kleinberger G, Yamanishi Y, Suárez-Calvet M, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 2014;6:243ra86.
116. Jiang T, Tan L, Zhu XC, et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014;39:2949-62.
117. Lee CYD, Daggett A, Gu X, et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s Disease models. Neuron 2018;97:1032-1048.e5.
118. Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med 2018;215:745-60.
119. Wang S, Mustafa M, Yuede CM, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med 2020;217:e20200785.
120. Ulrich JD, Finn MB, Wang Y, et al. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener 2014;9:20.
121. Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol Neurodegener 2018;13:29.
122. Ulland TK, Song WM, Huang SC, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s Disease. Cell 2017;170:649-663.e13.
123. Parhizkar S, Arzberger T, Brendel M, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 2019;22:191-204.
124. Jay TR, Miller CM, Cheng PJ, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med 2015;212:287-95.
125. Leyns CEG, Ulrich JD, Finn MB, et al. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A 2017;114:11524-9.
126. Bemiller SM, McCray TJ, Allan K, et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener 2017;12:74.
127. Sayed FA, Telpoukhovskaia M, Kodama L, et al. Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc Natl Acad Sci U S A 2018;115:10172-7.
128. Jiang T, Yu JT, Zhu XC, et al. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging 2014;35:1243-51.
129. Kleinberger G, Brendel M, Mracsko E, et al. The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J 2017;36:1837-53.
130. Bianchin MM, Capella HM, Chaves DL, et al. Nasu-Hakola Disease (Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy - PLOSL): A dementia associated with bone cystic lesions. from clinical to genetic and molecular aspects. Cell Mol Neurobiol 2004;24:1-24.
131. Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J Neurosci 2012;32:946-52.
132. Allendorf DH, Puigdellívol M, Brown GC. Activated microglia desialylate their surface, stimulating complement receptor 3-mediated phagocytosis of neurons. Glia 2020;68:989-98.
133. Gardai SJ, McPhillips KA, Frasch SC, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005;123:321-34.
134. Rauch JN, Luna G, Guzman E, et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020;580:381-5.
135. Holtzman DM, Pitas RE, Kilbridge J, et al. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci U S A 1995;92:9480-4.
136. Fuentealba RA, Liu Q, Zhang J, et al. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS One 2010;5:e11884.
137. Haure-Mirande JV, Wang M, Audrain M, et al. Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in cerebral Aβ amyloidosis mouse normalizes clinical phenotype and complement subnetwork molecular pathology without reducing Aβ burden. Mol Psychiatry 2019;24:431-46.
138. Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013;153:707-20.
139. Cong Q, Soteros BM, Wollet M, Kim JH, Sia GM. The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat Neurosci 2020;23:1067-78.
140. O’Brien JS, Sampson EL. Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res 1965;6:537-44.
141. Aureli M, Grassi S, Prioni S, Sonnino S, Prinetti A. Lipid membrane domains in the brain. Biochim Biophys Acta 2015;1851:1006-16.
142. Tufail Y, Cook D, Fourgeaud L, et al. Phosphatidylserine exposure controls viral innate immune responses by microglia. Neuron 2017;93:574-586.e8.
144. Cannon JP, O’Driscoll M, Litman GW. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 2012;64:39-47.
145. Shirotani K, Hori Y, Yoshizaki R, et al. Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Sci Rep 2019;9:7508.
146. Fourgeaud L, Través PG, Tufail Y, et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016;532:240-4.
148. Andreone BJ, Przybyla L, Llapashtica C, et al. Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat Neurosci 2020;23:927-38.
149. Marschallinger J, Iram T, Zardeneta M, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 2020;23:194-208.
151. Cunha C, Carvalho A, Esposito A, Bistoni F, Romani L. DAMP signaling in fungal infections and diseases. Front Immunol 2012;3:286.
152. Kumar DK, Choi SH, Washicosky KJ, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 2016;8:340ra72.
153. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, et al. Alzheimer’s Disease-associated β-Amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 2018;99:56-63.e3.
154. Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 2010;5:e9505.
155. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 2018;14:1602-14.
156. Pandey JP, Olsson J, Weidung B, et al. An Ig γ Marker genotype is a strong risk factor for Alzheimer Disease, independent of apolipoprotein E ε4 genotype. J Immunol 2020;205:1318-22.
157. Lövheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement 2015;11:593-9.
158. Warren-Gash C, Forbes HJ, Williamson E, et al. Human herpesvirus infections and dementia or mild cognitive impairment: a systematic review and meta-analysis. Sci Rep 2019;9:4743.
159. Readhead B, Haure-Mirande JV, Funk CC, et al. Multiscale analysis of independent alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018;99:64-82.e7.
160. Cairns DM, Rouleau N, Parker RN, Walsh KG, Gehrke L, Kaplan DL. A 3D human brain-like tissue model of herpes-induced Alzheimer’s disease. Sci Adv 2020;6:eaay8828.
161. Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett 2007;429:95-100.
162. McKenzie AT, Wang M, Hauberg ME, et al. Brain cell type specific gene expression and co-expression network architectures. Sci Rep 2018;8:8868.
163. Masuda T, Sankowski R, Staszewski O, Prinz M. Microglia heterogeneity in the single-cell era. Cell Rep 2020;30:1271-81.
164. van der Poel M, Ulas T, Mizee MR, et al. Transcriptional profiling of human microglia reveals grey-white matter heterogeneity and multiple sclerosis-associated changes. Nat Commun 2019;10:1139.
165. Sankowski R, Böttcher C, Masuda T, et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci 2019;22:2098-110.
166. Darmanis S, Sloan SA, Zhang Y, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A 2015;112:7285-90.
167. Zhang Y, Sloan SA, Clarke LE, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 2016;89:37-53.
168. Olah M, Patrick E, Villani AC, et al. A transcriptomic atlas of aged human microglia. Nat Commun 2018;9:539.
169. Forabosco P, Ramasamy A, Trabzuni D, et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging 2013;34:2699-714.
170. Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012;489:391-9.
171. Del-Aguila JL, Li Z, Dube U, et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimers Res Ther 2019;11:71.
172. Galatro TF, Holtman IR, Lerario AM, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci 2017;20:1162-71.
173. Mrdjen D, Pavlovic A, Hartmann FJ, et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018;48:380-395.e6.
174. Van Hove H, Martens L, Scheyltjens I, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 2019;22:1021-35.
175. Jordão MJC, Sankowski R, Brendecke SM, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019;363:eaat7554.
176. Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014;34:11929-47.
177. Tasic B, Menon V, Nguyen TN, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 2016;19:335-46.
178. Zeisel A, Muñoz-Manchado AB, Codeluppi S, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015;347:1138-42.
179. Ximerakis M, Lipnick SL, Innes BT, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci 2019;22:1696-708.
180. Hammond TR, Dufort C, Dissing-Olesen L, et al. Single-cell RNA Sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019;50:253-271.e6.
181. Saunders A, Macosko EZ, Wysoker A, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 2018;174:1015-1030.e16.
182. Tay TL, Sagar, Dautzenberg J, Grün D, Prinz M. Unique microglia recovery population revealed by single-cell RNAseq following neurodegeneration. Acta Neuropathol Commun 2018;6:87.
183. Holtman IR, Raj DD, Miller JA, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun 2015;3:31.
184. Pedersen NL, Gatz M, Berg S, Johansson B. How heritable is Alzheimer’s disease late in life? Findings from Swedish twins. Ann Neurol 2004;55:180-5.
185. Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168-74.
186. Gatz M, Pedersen NL, Berg S, et al. Heritability for Alzheimer’s disease: the study of dementia in Swedish twins. J Gerontol A Biol Sci Med Sci 1997;52:M117-25.
187. Bergem AL, Engedal K, Kringlen E. The role of heredity in late-onset Alzheimer disease and vascular dementia. A twin study. Arch Gen Psychiatry 1997;54:264-70.
188. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993;261:921-3.
189. Harold D, Abraham R, Hollingworth P, et al. Erratum: Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2013;45:712.
190. Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013;45:1452-8.
191. Alzforum. AlzGene - Overview of all published large-scale and genome-wide association studies in AD. Available from: http://www.alzgene.org/largescale.asp. [Last accessed on 29 Jan 2021].
192. Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019;51:414-30.
193. de Rojas I, Moreno-Grau S, Tesi N, et al. Common variants in Alzheimer’s disease: novel association of six genetic variants with AD and risk stratification by polygenic risk scores. medRxiv 2020:19012021.
194. Moreno-Grau S, de Rojas I, Hernández I, et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: The GR@ACE project. Alzheimers Dement 2019;15:1333-47.
195. Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 2019;51:404-13.
196. Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry 2018;8:99.
197. Sims R, van der Lee SJ, Naj AC, et al; ARUK Consortium., GERAD/PERADES, CHARGE, ADGC, EADI. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 2017;49:1373-84.
198. Holstege H, Hulsman M, Charbonnier C, et al. Exome sequencing identifies novel AD-associated genes. medRxiv 2020:20159251.
199. Guerreiro R, Wojtas A, Bras J, et al; Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117-27.
200. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s Disease. N Engl J Med 2013;368:107-16.
201. Fernández MV, Budde J, Del-Aguila JL, et al; NIA-LOAD family study group., NCRAD. Evaluation of Gene-based family-based methods to detect novel genes associated with familial late onset Alzheimer Disease. Front Neurosci 2018;12:209.
202. Nott A, Holtman IR, Coufal NG, et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 2019;366:1134-9.
203. Novikova G, Kapoor M, Julia TCW, et al. Integration of Alzheimer’s disease genetics and myeloid genomics reveals novel disease risk mechanisms. BioRxiv 2019; doi: 10.1101/694281.
204. Amlie-Wolf A, Tang M, Way J, et al. Inferring the molecular mechanisms of noncoding Alzheimer’s Disease-associated genetic variants. J Alzheimers Dis 2019;72:301-18.
205. Jones L, Holmans PA, Hamshere ML, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One 2010;5:e13950.
206. Galatro TF, Vainchtein ID, Brouwer N, Boddeke EWGM, Eggen BJL. Isolation of microglia and immune infiltrates from mouse and primate central nervous system. In: Clausen BE, Laman JD, editors. Inflammation. New York: Springer; 2017. pp. 333-42.
207. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248-9.
208. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc 2016;11:1-9.
209. Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016;99:877-85.
210. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011;39:e118.
211. Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015;24:2125-37.
212. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012;7:e46688.
213. Park JS, Ji IJ, Kim DH, An HJ, Yoon SY. The Alzheimer’s Disease-associated R47H variant of TREM2 has an altered glycosylation pattern and protein stability. Front Neurosci 2016;10:618.
214. Thornton P, Sevalle J, Deery MJ, et al. TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol Med 2017;9:1366-78.
215. Jiang T, Hou JK, Gao Q, et al. TREM2 p.H157Y variant and the risk of Alzheimer’s Disease: a meta-analysis involving 14,510 subjects. Curr Neurovasc Res 2016;13:318-20.
216. Kober DL, Alexander-Brett JM, Karch CM, et al. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 2016;5:e20391.
217. Sudom A, Talreja S, Danao J, et al. Molecular basis for the loss-of-function effects of the Alzheimer’s disease-associated R47H variant of the immune receptor TREM2. J Biol Chem 2018;293:12634-46.
218. Song W, Hooli B, Mullin K, et al. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement 2017;13:381-7.
219. Paloneva J, Manninen T, Christman G, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 2002;71:656-62.
220. Raj T, Ryan KJ, Replogle JM, et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet 2014;23:2729-36.
221. Estus S, Shaw BC, Devanney N, Katsumata Y, Press EE, Fardo DW. Evaluation of CD33 as a genetic risk factor for Alzheimer’s disease. Acta Neuropathol 2019;138:187-99.
222. Cheng Q, Danao J, Talreja S, et al. TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer’s disease variant Trem2R47H on murine myeloid cell function. J Biol Chem 2018;293:12620-33.
223. Schlepckow K, Monroe KM, Kleinberger G, et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med 2020;12:e11227.
224. Hampe W, Riedel IB, Lintzel J, Bader CO, Franke I, Schaller HC. Ectodomain shedding, translocation and synthesis of SorLA are stimulated by its ligand head activator. J Cell Sci 2000;113:4475-85.
225. Hermey G, Sjøgaard SS, Petersen CM, Nykjaer A, Gliemann J. Tumour necrosis factor alpha-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem J 2006;395:285-93.
226. Böhm C, Seibel NM, Henkel B, Steiner H, Haass C, Hampe W. SorLA signaling by regulated intramembrane proteolysis. J Biol Chem 2006;281:14547-53.
227. Satoh T, Arii J, Suenaga T, et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008;132:935-44.
228. Rathore N, Ramani SR, Pantua H, et al. Paired Immunoglobulin-like Type 2 Receptor Alpha G78R variant alters ligand binding and confers protection to Alzheimer’s disease. PLoS Genet 2018;14:e1007427.
229. Chierico L, Joseph AS, Lewis AL, Battaglia G. Live cell imaging of membrane/cytoskeleton interactions and membrane topology. Sci Rep 2014;4:6056.
230. Lee K, Gallop JL, Rambani K, Kirschner MW. Self-assembly of filopodia-like structures on supported lipid bilayers. Science 2010;329:1341-5.
231. Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal 2010;3:ra38.
232. Li C, Zhao B, Lin C, Gong Z, An X. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-κB signaling. Cell Biol Int 2019;43:360-72.
233. Bernier LP, Bohlen CJ, York EM, et al. Nanoscale surveillance of the brain by microglia via cAMP-regulated filopodia. Cell Rep 2019;27:2895-2908.e4.
234. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 2008;9:690-701.
235. Ueno T, Falkenburger BH, Pohlmeyer C, Inoue T. Triggering actin comets versus membrane ruffles: distinctive effects of phosphoinositides on actin reorganization. Sci Signal 2011;4:ra87.
236. Oikawa T, Yamaguchi H, Itoh T, et al. PtdIns(3,4,5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nat Cell Biol 2004;6:420-6.
237. Phillips AEM, Villegas-Llerena C, Piers TM, Cosker K, Hardy J, Pocock JM. Loss of function of TREM2 results in cytoskeletal malfunction in microglia. J Neurol Neurobiol 2018;4.
238. Cella M, Buonsanti C, Strader C, Kondo T, Salmaggi A, Colonna M. Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J Exp Med 2003;198:645-51.
239. Litschko C, Linkner J, Brühmann S, et al. Differential functions of WAVE regulatory complex subunits in the regulation of actin-driven processes. Eur J Cell Biol 2017;96:715-27.
240. Chen XJ, Squarr AJ, Stephan R, et al. Ena/VASP proteins cooperate with the WAVE complex to regulate the actin cytoskeleton. Dev Cell 2014;30:569-84.
241. Havrylenko S, Noguera P, Abou-Ghali M, et al. WAVE binds Ena/VASP for enhanced Arp2/3 complex-based actin assembly. Mol Biol Cell 2015;26:55-65.
242. Moraes L, Zanchin NIT, Cerutti JM. ABI3, a component of the WAVE2 complex, is potentially regulated by PI3K/AKT pathway. Oncotarget 2017;8:67769-81.
243. Haenseler W, Sansom SN, Buchrieser J, et al. A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Reports 2017;8:1727-42.
244. Buchrieser J, James W, Moore MD. Human induced pluripotent stem cell-derived macrophages share ontogeny with MYB-Independent tissue-resident macrophages. Stem Cell Reports 2017;8:334-45.
245. Abud EM, Ramirez RN, Martinez ES, et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 2017;94:278-293.e9.
246. Muffat J, Li Y, Yuan B, et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med 2016;22:1358-67.
247. Pandya H, Shen MJ, Ichikawa DM, et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci 2017;20:753-9.
248. Takata K, Kozaki T, Lee CZW, et al. Induced-pluripotent-stem-cell-derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity 2017;47:183-198.e6.
249. Brownjohn PW, Smith J, Solanki R, et al. Functional studies of missense TREM2 mutations in human stem cell-derived microglia. Stem Cell Reports 2018;10:1294-307.
250. Hooper C, Taylor DL, Pocock JM. Pure albumin is a potent trigger of calcium signalling and proliferation in microglia but not macrophages or astrocytes. J Neurochem 2005;92:1363-76.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Hodges AK, Piers TM, Collier D, Cousins O, Pocock JM. Pathways linking Alzheimer's disease risk genes expressed highly in microglia. Neurosciences 2021;8:245. http://dx.doi.org/10.20517/2347-8659.2020.60
AMA Style
Hodges AK, Piers TM, Collier D, Cousins O, Pocock JM. Pathways linking Alzheimer's disease risk genes expressed highly in microglia. Neuroimmunology and Neuroinflammation. 2021; 8: 245. http://dx.doi.org/10.20517/2347-8659.2020.60
Chicago/Turabian Style
Hodges, Angela K., Thomas M. Piers, David Collier, Oliver Cousins, Jennifer M. Pocock. 2021. "Pathways linking Alzheimer's disease risk genes expressed highly in microglia" Neuroimmunology and Neuroinflammation. 8: 245. http://dx.doi.org/10.20517/2347-8659.2020.60
ACS Style
Hodges, AK.; Piers TM.; Collier D.; Cousins O.; Pocock JM. Pathways linking Alzheimer's disease risk genes expressed highly in microglia. Neurosciences. 2021, 8, 245. http://dx.doi.org/10.20517/2347-8659.2020.60
About This Article
Copyright

Data & Comments
Data


 Cite This Article 34 clicks
Cite This Article 34 clicks

 Like This Article 19
likes
Like This Article 19
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.