
Resolution of inflammation and repair after ischemic brain injury

Abstract
After ischemic stroke, proinflammatory molecules known as danger-associated molecular patterns (DAMPs) originating from damaged brain cells recruit and activate immune cells (neutrophils, macrophages, lymphocytes) further eliciting innate and adaptive immunity. During the acute phase from day 1 to day 3 of the stroke onset, macrophages play a major role in the progression of inflammation, promoting the destruction of brain tissue. During the recovery phase, from day 3~4 to day 7 after stroke onset, infiltrating macrophages switch to repairing macrophages, which clear the DAMPs and promote tissue repair by producing neurotrophic factors. Adaptive immunity during the late or chronic phase (> day 7) of stroke has not been well investigated. Recent studies have also indicated that antigen-specific T cells, especially regulatory T cells (Tregs), play major roles in neural repair. This review focuses mainly on the resolution of inflammation and tissue repair by macrophages and Tregs.
Keywords
Introduction
Ischemic cerebral infarction accounts for 70% to 80% of all strokes, which is the leading cause of severe neuropathy, disability and bedriddenness[1]. Ischemic stroke causes the death of nerve cells as well as destruction of neuronal circuits, which leads to movement disorders, higher brain dysfunction, and sensory disturbance. Ischemic damages of the brain tissue further induce cerebral edema and inflammation, which exacerbates the functional prognosis and symptoms of stroke. Although administration of tissue-plasminogen activator reduces ischemic neural damages, this treatment should be instituted within 4.5 h of stroke onset[2]. After this acute stage, no therapeutic drugs should be administered during the late stage of stroke, and following this short period, rehabilitation is the only modality of treatment for functional recovery at present.
Sterile inflammation initially leads to tissue damages[3]. Likewise, in ischemic stroke, brain inflammation causes neural cell death and has been considered to be an attractive target for reducing brain damages not only in experimental rodent models but also in human patients[4-6]. As inflammation occurs within a few days after stroke onset, innate immunity, in which microglia, macrophages, neutrophils, and γδT cells play major role, has been thought to account for neuroinflammation after stroke, where such inflammation disappears after 1 week of stroke onset [Figure 1]. To date, only a small number of studies have investigated the adaptive immunity in stroke, which usually occurs over a week after the onset of the disease or infection[7,8]. However, our group and others have discovered an accumulation of lymphocytes including regulatory T cells (Tregs) in the brain at the chronic phase (more than 2 weeks) after stroke onset. This process has been shown to be involved in neural repair rather than progression of the disease[9-13].
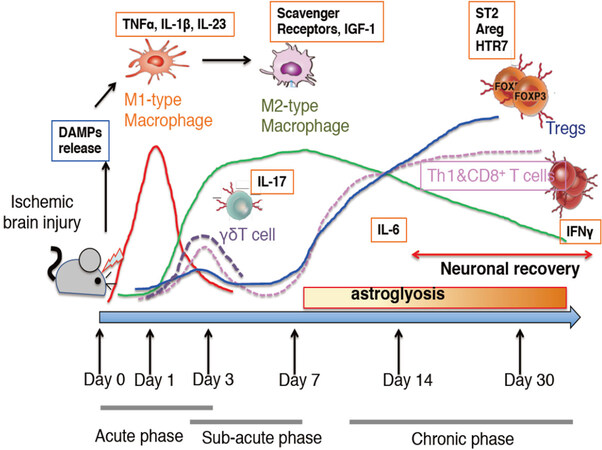
Figure 1. Schematic viewing a time-dependent recruitment of various inflammatory cells into the brain following cerebral ischemia in mice. In this review, we refer to day 1-3 after stroke onset as the acute phase, for days 3-7 being subacute phase, and period after 2 weeks being chronic phase. This figure illustrates the conceptual changes in the population of immune cells, thus the numbers of each immune cell may not necessarily be accurate. Red line; M1 type inflammatory macrophages, green line; M2 type macrophages, gray broken line; γδT cells, pink broken line; Th1 cells and CD8+ T cells, blue line; regulatory T cells (Tregs). IL: interleukin; IGF: insulin growth factor
In this mini-review article, we will focus on the resolution of inflammation and tissue repair of the brain after ischemic stroke.
Inflammation cascade after stroke
In the very acute stage (< 24 h) after stroke onset, the major player involved is infiltrated macrophage, which can be differentiated from bone marrow-derived monocytes. Macrophages are activated by extracellular molecules known as danger-associated molecular patterns (DAMPs), which are released from damaged and dead cells. Infiltrated macrophages at this stage are highly pro-inflammatory and produce cytokines, chemokines and mediators which exacerbate ischemic encephalopathy, leading to dysfunction of the blood brain barrier (BBB)[4,14] [Figures 1 and 2]. HMGB1, S100A8 and S100A9, and peroxiredoxin-family proteins are major DAMPs which activate infiltrated macrophages through toll-like receptor (TLR)-2 and TLR-4[15].
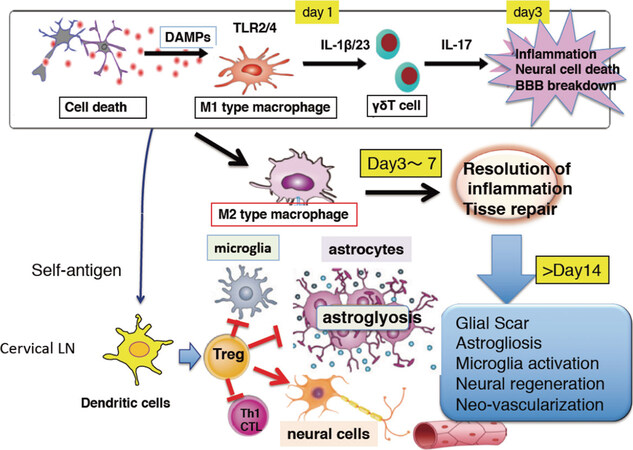
Figure 2. Schematic view of the role of immune cell types in cytokine production, neuroinflammation, and tissue repair. In the early stage of around 24 h, macrophages infiltrate the injured brain and are activated through the TLR2/4 stimulation by molecules known as danger-associated molecular patterns (DAMPs) from necrotic cells. Thereafter, infiltrated macrophages produce inflammatory cytokines and mediators which increase probability of ischemic encephalopathy and blood brain barrier (BBB) destruction. At this stage, macrophages become M1-type inflammatory macrophages. Further on, IL-23 and IL-1β from inflammatory macrophages stimulate IL-17 production from infiltrated γδT cells. After day 3, macrophages are converted into M2-type repairing macrophages which are involved in clearing DAMPs, resolving inflammation, and tissue repair by producing neurotrophic factors such as IGF-1. During the chronic phase, a massive T-cell infiltration occurs. Brain Tregs are attracted via chemokines and proliferate in the cervical lymph node (LN) and the brain. Brain Tregs may thereafter interact with various brain cells including that of microglia, astrocytes, endothelial cells, and neural cells promoting neural cell recovery in th process. IL: interleukin; IGF: insulin growth factor
Among inflammatory cytokines released from macrophages, TNFα, interleukin (IL)-1β, IL-23 have been shown to contribute significantly to brain damages and neural dysfunctions[16-19]. In particular, IL-1β and IL-23 induce IL-17 production from infiltrated γδT cells, further promote inflammation, BBB breakdown, and neuronal damage[19-21]. IL-17 is a cytokine which has been shown to play important pathological roles not only in ischemic stroke, but also in various neuroinflammation including neurodegenerative diseases[22-24]. The inflammasomes, which are necessary for mature IL-1β release, have also been described to cause deterioration in infarct volume resulting in neural defects in stroke patients. Inhibition of inflammasome is shown to be effective for reducing neuroinflammation and subsequently reducing infarct volume increase[20,25].
Resolution mechanism of inflammation after stroke
After 3-4 days post-ischemic stroke onset, inflammatory macrophages termed M1 type macrophages, are converted into repairing macrophages or M2-type macrophages. Repairing macrophages play a role in scavanging tissue debris and necrotic cells, futher supporting neural repair by releasing neurotrophic factors including insulin growth factor (IGF)-I[26][Figure 2]. It has been suggested by some studies that IL-10 and TGF-β from M2-type macrophages and microglia, promoting the resolution of inflammation[27]. Is is not clear whether the same M1 macrophages may convert to M2 type, or M2 macrophages replace M1 type macrophages although an imaging study of infiltrated macrophages in experimental autoimmune encephalomyelitis (EAE) model revealed that single macrophage changes its phenotype from M1 to M2[28]. For clearance of DAMPs, Msr1 (macrophage scavenger receptor-1 or what is known as CD204 or SCARA1) was identified as a major scavanger receptor[29,30]. Msr1 promoter was described to be activated by Maf-b, and an RAR agonist, Am80, upregulating MAF-B expression, therefore promoting Msr1 expression and clearance of DAMPs which ultimately facilitates neurological recovery[29]. Am80 has also been shown to be neuroprotective by activating the PI3-kinase/Akt pathway[31]. Msr1 has been shown in several studies to clear various neurotoxic molecules including amyloid-β thus playing an important neuroprotective role[32,33]. Mannose receptors on infiltrating macrophages have also been reported to be involved in the clearance of DAMPs in focal cortical ischemia[34].
The early activation of microglia in the post-ischemic brain was demonstrated to be neuroprotective by regulating neuronal Ca2+ overload and spread of depolarization. Pharmacological ablation of microglia results in infract size increase and dysregulation of neuronal circuit, while microglia repopulation reverses these effects[35]. The pro-resolving mediators including protectins and resolvins, which have been shown to be neuroprotective. Resolvins reduce neural damage through suppression of leukocyte infiltration, IL-1β expression, and NF-κB activation[36]. Neuroprotectin-D1 similarly reduces infarct volume and diminishes disease burden[37]. LXA4 had also been reported to be neuroprotective by virtue of mitigating astrogliosis, IL-1β, TNFα expression, and neutrophil infiltration. Additionally, it also converts phenotypes of monocytes from inflammatory to an anti-inflammatory and serves functionally to repair tissues[38]. However, the mechanism of resolution (or suppression) of inflammation by microglia and these lipid-mediators remains to be described.
Lack of CCR5 expression has been reported to increase the severity of ischemic brain injury[39]. CCR5 is uniquely expressed in cortical neurons within the damaged brain[40]. CCR5 antagonists accelerate recovery from neurological and cognitive dysfunction. Although various roles of CCR5 in neurons have been reported, the inhibition of CCR5 has been found to suppress astrocyte reactivity and macrophage recruitment[40,41]. Nevertheless, another study has revealed the pathogenic role of CCR5 in cerebral ischemia[42], suggesting that various types of cells may express CCR5 and contribute to both neuronal inflammation and tissue repair of the ischemic brain.
Role of microglia in resolution of inflammation and neural repair
Microglia have been shown to play important roles in neural inflammation, resolution of inflammation and clearance of dead cells in the brain[43]. Since major sources of IL-1β and IL-23 is infiltrated M1 type macrophages originate during the acute phase of inflammation after stroke model[14,20], the contribution of microglia to severe inflammation post stroke is not very clear. Depletion of microglia leads to aggravated neuronal damage and apoptosis after ischemic brain injury, suggesting that microglia plays an important role in neuroprotection[44]. Mechanistically, microglia senses damaged cells through the purinergic receptors (P2X4R, P2X6R, P2X12R) which respond to adenosine triphosphate released from dead cells. The activation of the purinergic receptors initiates neuroprotective responses rather than destructive inflammation through the recruitment of microglia to the point of injury[45,46]. Microglia also express clearance receptors such as Msr1, complement receptors, and receptors for apoptotic cells[32,47]. A recent study shows beneficial effects of repopulating microglia which support adult neurogenesis by augmenting the survival of newborn neurons in traumatic brain injury thorough IL-6 trans-signaling pathways[48]. IGF-1 produced by microglia has been found to be neuroprotective[49]. However, the significance of IGF-1 in neuroinflammation and neuroprotectin remain controversial[50].
Role of lymphocytes in post-ischemic brain inflammation
It has been reported that lymphocytes including T cells and B cells play various roles in the pathophy-siology of stroke[51]. B cells and immunoglobulins are detected within and around the stroke core in a subgroup of stroke and dementia patients, and also in a murine experimental stroke model. Several studies suggest that post-stroke cognitive impairment has been associated with B cell activation and auto-antibody production[52]. Nevertheless, the specific roles of B cells and/or antibodies in neurological deficits and inflammation after ischemic brain injury remain uncertain[53]. Plasma cells in the central nervous system (CNS) of mice with EAE have been shown to originate in the gut and produce IgA, which confers resistance to mice to the effector stage of EAE through the production of IL-10[54]. Interestingly, stroke patients demonstrate a type of auto-immunoreactivity to brain antigens[55].
T cells have been more intensively investigated than B cells. This is mostly because various cytokines, such as IL-10, IL-17, IL-21, IFN-γ, and TNF-α produced from CD4+T cells and/or γδT cells, affect and regulate glial cells, endothelial cells, neural cells, and various immune cells[56,57]. IL-21 is predominantly produced from CD4+T cells, but the role of this particular cytokine in stroke is controversial. In mice, a locus on distal chromosome 7 has been described to contribute variations in post-ischemic cerebral infarct volume, and the IL-21 receptor has been identified as a strong candidate which functions in a neuroprotective manner[58]. However, another study suggested that IL-21 promotes brain injury after stroke in mice[57], thus further research is necessary to clarify the role of IL-21 in brain injury.
The route of infiltration of T cells to the brain is not described clearly. A recent study shows that T cells specifically accumulate within the peri-infarct cortex after stroke and that the ipsilateral choroid plexus plays a key cerebral invasion route for T cells[59]. This study suggests that the CCR2-ligand gradient between cortex and choroid plexus serves as the potential driving force for T cell invasion.
In the experimental cerebral ischemia model, infiltration of subsets of T cells occurs at various time points [Figures 1 and 2]. Many reports indicate that T cells promote brain damage at the early phase of stroke[19,60]. In humans, FTY720 (fingolimod) treatment within 72 h post stroke onset blocks the infiltration of pathogenic T cells into the brain, effectively ameliorating neurological symptoms in the patient[61]. CD8+ T cells may infiltrate within several hours after stroke onset[61]. Nerve-damaging substances such as granzymes and perforin from CD8+T cells can exacerbate the infarction[62]. γδT cells increase immediately after stroke onset and are present in the brain parenchyma accompanied with BBB breakdown. On day 3 after stroke onset, the number of γδT cells reaches its maximum concentrations[19,21]. IL-17 produced from γδT cells promotes neural cell damages in the ischemic penumbra region[19,21,63]. Taken together, γδT cells depletion well as anti-IL-17 neutralizing antibody are shown to suppress ischemic brain injury[19,64]. CD4+T cells and NKT cells infiltrate the brain after 24 h of ischemic stroke[65]. These reports indicate that T cell could serve as a therapeutic target for stroke. However, we should remain cautious that within one week after stroke onset, the number of T cells in the brain is small and represents only a small fraction of infiltrated mononuclear cells[19][Figure 1].
It has been reported that during the acute phase of stroke, Tregs infiltrate the brain, suppressing neuroinflammation, therby reducing the severity of ischemic brain injury[60,66,67]. However, the significance of Tregs in brain injury has become controversial[68,69]. Importantly, the number of Tregs in the brain at this stage is extremely low (less than 100 cells/brain in mice), and antigen-specific activation and proliferation of Tregs may not occur in such a short period (within 3 days), since it usually takes more than a week to raise adaptive immunity in the host [Figures 1 and 2]. Thus, bystander effects, such as paracrine effects of IL-10, may explain an anti-inflammatory role of Tregs at the acute phase.
Accumulation of brain Tregs at the chronic phase of stroke
It is thought that inflammation no longer plays an important role in neural damage and recovery at the chronic phase of stroke (> 7 days after stroke onset). Inflammation is not clearly obvious at this stage. However, compared to the acute phase, Tregs as well as other lymphocytes have been shown to accumulate in substantial quantities in the brain at the chronic phase of the experimental stroke model[9-13] [Figures 1 and 2]. Infiltration of Tregs proceeds with slightly delayed kinetics compared with that of other T cells. Tregs may also infiltrate the spinal cord parenchyma during the subacute to chronic phases in the spinal cord injury model[70]. Tregs consist of approximately 50% of CD4+ T cells, and localize within and around the cerebral infarction lesion. Outside the infarct core area, Tregs remain in close proximity to scar-forming astrocytes and neuronal cells. Treg fractions in the brain are extremely higher that those in other lymphoid organs such as the spleen and lymph nodes. Since CD8+ T cells are also present, about 1/4 to 1/5 of T cells in the brain are calculated as Tregs.
To determine the role of T cells in ischemic brain injury, mice were treated with FTY720 or anti-CD4 antibody during the chronic phase after stroke. Tregs can also be depleted by the use of Foxp3-diphtheria toxin receptor (DTR) mice, where DTR is specifically expressed on Tregs[71]. These treatments drastically reduce the number of CD4+T cells including Tregs in the brain, delaying neurological recovery. These data indicate that brain Tregs at the chronic phase are important for suppressing neurological symptoms[9]. Stubbe et al.[10] observed no changes regarding neurologic outcome if they depleted Tregs through the use of anti-CD25 antibody[10]. Anti-CD25 antibody, however, may not be able to completely deplete Tregs, possibly depleting pathogenic T cells as well[72]. Other studies have also shown that brain Tregs play neuroprotective roles during the late stage of stroke and spinal cord injury models[12,73].
Characterization of brain Tregs
Tregs consist of approximately 10% of CD4+ T cells, located within most lymphoid organs and blood, moving to specific sites of inflammation after immunization. In addition, Tregs have recently been discovered in various tissues besides lymphoid tissues, in steady state conditions as well as during injury. These tissue-residing Tregs are now termed “tissue Tregs”, which have a limited TCR repertoire and recognize the self-antigen characteristically expressed in each tissue. Such tissue Tregs exist in fats, muscles, skin, lungs, and intestines, exhibiting similar phenotypes among organs, but are quite different from those of lymphoid tissue[74-78]. The features common to various tissue Tregs are high expressions of Il10, Areg (amphiregulin), Klrg1, Tigit, Il1rl1 (encoding ST2, IL-33 receptor), Ctla4, Irf4, Batf, and Gata3 and low expressions of Bcl2, Tcf7, and Lef1 compared with lymphatic Tregs[74,79]. BATF is shown to be an important regulator for Tregs to accumulate preferentially in several tissues[80]. In addition to the common genes expressed in various tissue Tregs, unique tissue-specific genes are also found in tissue Tregs namely Pparg in fat Tregs. The microenvironment of each organ appears to determine the tissue-specific phenotypes of tissue Tregs.
Like other tissue Tregs, brain Tregs express Helios, which suggests that brain Tregs are derived from the thymus embryonically. Brain Tregs possess a unique TCR repertoire and express high levels of CTLA-4, PD-1, Areg, KLRG1, and ST2, indicating that the brain Tregs share common features of tissue Tregs.
Molecules deeply involved in Treg-mediated neural recovery
Chemokine receptors
Tissue Tregs express unique chemokine receptors in each organ. Brain Tregs express specific chemokine receptors including CCR6 and CCR8, and their ligands. CCL20 and CCL1 in particular are highly expressed in the cerebral infarct area. Intra-ventricular injection of CCL1 and CCL20 has been reported to increase the number of Tregs, resulting in improvement of neurological recovery[9].
IL-33
Of note, IL-33 promotes tissue recovery after CNS injury[81] and up-regulates M2 type macrophage-related genes[82]. Since many reports suggest that IL-33 induces the expansion of Tregs in the brain[9,12,83], it is highly possible that brain Tregs are involved in the beneficial effects on CNS damage[12,84]. In the skeletal muscle injury model, local mesenchymal stromal cells express the receptor for the calcitonin-gene-related peptide (CGRP), producing IL-33 in response to CGRP, which further promotes accumulation of Tregs and muscle tissue repair[78]. IL-33-expressing cells in the brain consist of astrocytes and oligodendrocytes[9]. Higher serum IL-33 levels in acute ischemic stroke patients correlated positively with better prognosis, as compared with those with lower IL-33 levels. These patients presented with poorer outcome[85] suggesting that IL-33 is protective to stroke.
Serotonin receptor
As earlier described, tissue Tregs express a common set of genes among various tissue Tregs, while each organ-specific Treg expresses tissue-specific genes. Unlike other tissue Tregs, brain Tregs express several unique CNS-related genes. For example, brain Tregs express serotonin receptor 7 (Htr 7), which increases cellular cAMP[86]. It has been acknowledged that cAMP promotes the Treg proliferation and thereby potentiating Treg functions[87]. Serotonin was reported to decrease Th1/Th17 cytokines, but increased Treg population in multiple sclerosis (MS) patients[88]. Serotonin further activates Tregs from the ischemic brain in vitro in an Htr7-dependent manner. The administration of serotonin or a selective serotonin reuptake inhibitor (SSRI) thus increases the number of brain Tregs in the chronic phase after stroke onset, improving neurological symptoms[9]. Many reports suggest that SSRI ameliorates neurological symptoms after stroke onset[89,90], although some studies did not prove that functional recovery improved[91]. It is highly likely that brain Tregs work on neuronal repair in human stroke patients.
Amphiregulin
Hypertrophic astrocytes exhibit increased Ca2+ signaling, which leads to the increased expression of pro-inflammatory cytokines (IL-1β, IL-6) and chemokines (CCL3, CCL5) expression, promoting the formation of glial scars[92]. Amphiregulin (Areg) is also known to suppress the production of inflammatory cytokines, including IL-6 and TNFα, in several inflammatory diseases[93].
Areg from brain Tregs has been described to suppress the excessive activation of astrocytes, so-called astrogliosis or reactive astrocytes, which is then reported to lead to a delay in the recovery from ischemic stroke or spinal cord injury[94]. Although astrogliosis would be necessary for forming scars in order to demarcate the ischemic regions from the surrounding healthy tissue, excessively activated astrocytes can produce neurotoxic factors, resulting in neural cell damage[95]. Areg further suppresses apoptosis of neurons by suppressing excessive astrocyte activation[9]. The molecular mechanism of suppression of astrocyte activation by Areg has not been completely elucidated. Among inflammatory cytokines, IL-6 is important for astrocyte activation[96]. Since Areg suppresses IL-6 expression in microglia and astrocytes in vivo and in vitro[9], suppression of IL-6 by Areg may play a key role in astrocyte regulation. In addition, Areg has been postulated to be directly involved in the proliferation of neural stem cells[97].
Enkephalin
Prepro-enkephalin is produced in Regulatory T cells[98]. Pharmacologically active encephalin has been reported to be therapeutic for stroke[99]. Opioid growth factor ([Met(5)]-enkephalin) is also neuroprotective in the murine EAE model[100]. However, some studies indicate that preproenkephalin accelerates the generation of autoimmune IFN-γ-producing T cells and exacerbates EAE[101].
PPARγ
PPARγ levels are high in brain Tregs. PPARγ agonists have been reported to increase accumulation of adipose-tissue Tregs and improve insulin sensitivity[102] In particular, PPARγ agonists have a protective effect against various types of injury to the brain[103]. Thus, it is possible that PPARγ is involved in Treg expansion in the brain, which should be further investigated.
Tregs and human CNS inflammatory diseases
Accumulation of Tregs in the human brain of ischemic stroke patients has not been clearly shown. However, a correlation between peripheral blood Treg/Th17 ratio or IL-17/IL-10 levels and stroke prognosis has been reported in human stroke[104-107]. An inverse correlation between the number of Tregs in the peripheral blood and the severity of stroke has also been reported in patients[13,108].
It has been established that neural inflammation plays important roles not only in cerebral infarction, but also in various types of damage to cerebrospinal tissues. These include spinal cord injury, autoimmune diseases such as multiple sclerosis, and in neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. Innate and adaptive immunity may be involved in these neural inflammations. Although Tregs have been shown to infiltrate and accumulate within the CNS[109] of neuroinflammatory diseases, role of Tregs in such diseases have not been well characterized. Since Tregs of MS patients have been shown to proliferate by serotonin stimulation[88], Tregs in the CNS diseases may be similar to the brain Tregs that have been characterized in a murine ischemic stroke model.
Conclusion
In summary, brain macrophages as well as brain Tregs play important roles in the resolution of inflammation and neural recovery. The conversion mechanism from inflammatory macrophages to tissue-repair macrophages and the mechanism of expansion of brain Tregs by recognizing self-antigens in the cervical LN and the brain remain to be described. Neuroprotective factors such as IGF-1 from macrophages and Areg produced by brain Tregs not only suppress excessive activation of microglia and astrocytes, but may also promote neural cell survival and neural stem cell recruitment [Figure 2][110,111]. The molecular mechanisms whereby macrophages and Tregs acquire brain-specific characteristics, including Maf-b/scavenger receptor expression and serotonin receptor expression, respectively, remain to be clarified. Such mechanisms could be used for increasing Tregs in the brain. Identification of brain factors and self-antigens for brain-specific macrophages and Tregs may facilitate the development of therapies for not only cerebral infarction but also other central nervous system diseases. Adoptive transfer of mesenchymal stem cells to the brain has been proposed to treat stroke patients[112]. Similarly autologous Tregs transfer into the brain is also possible for the treatment of cerebral inflammation. It is also important to define the role of brain Tregs in other neurodegenerative disorders and neuroinflammatory diseases.
Declarations
Authors’ contributionsSearched literatures and wrote the manuscript: Yoshimura A, Ito M
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by JSPS KAKENHI (S) JP17H06175, Challenging Research (P) JP18H05376, and AMED-CREST JP20gm1110009 to Yoshimura A, and JSPS KAKENHI 17K15667, 19H04817, and 19K16618, AMED-PRIME 20gm6210012 to Ito M and by the Tomizawa Jun-ichi & Keiko Fund of Molecular Biology Society of Japan for Young Scientists, a Research Grant for Young Investigators by The Mitsubishi Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Takeda Science Foundation, the Uehara Memorial Foundation, the Naito Memorial Foundation, the Kanae Foundation, the SENSHIN Medical Research Foundation, the Astellas Foundation for Research on Metabolic Disorders, an Inoue Research Award, a Life Science Research Award, and Keio Gijuku Academic Developmental Funds.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Ojaghihaghighi S, Vahdati SS, Mikaeilpour A, Ramouz A. Comparison of neurological clinical manifestation in patients with hemorrhagic and ischemic stroke. World J Emerg Med 2017;8:34-8.
3. Shichita T, Ago T, Kamouchi M, Kitazono T, Yoshimura A, et al. Novel therapeutic strategies targeting innate immune responses and early inflammation after stroke. J Neurochem 2012;123 Suppl 2:29-38.
4. Shichita T, Ito M, Yoshimura A. Post-ischemic inflammation regulates neural damage and protection. Front Cell Neurosci 2014;8:319.
5. Jiang C, Kong W, Wang Y, Ziai W, Yang Q, et al. Changes in the cellular immune system and circulating inflammatory markers of stroke patients. Oncotarget 2017;8:3553-67.
6. Dokalis N, Prinz M. Resolution of neuroinflammation: mechanisms and potential therapeutic option. Semin Immunopathol 2019;41:699-709.
7. Javidi E, Magnus T. Autoimmunity after ischemic stroke and brain injury. Front Immunol 2019;10:686.
8. Malone K, Amu S, Moore AC, Waeber C. The immune system and stroke: from current targets to future therapy. Immunol Cell Biol 2019;97:5-16.
9. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019;565:246-50.
10. Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab 2013;33:37-47.
11. Chen C, Chencheng Z, Cuiying L, Xiaokun G. Plasmacytoid dendritic cells protect against middle cerebral artery occlusion induced brain injury by priming regulatory T cells. Front Cell Neurosci 2020;14:8.
12. Liu X, Hu R, Pei L, Si P, Wang C, et al. Regulatory T cell is critical for interleukin-33-mediated neuroprotection against stroke. Exp Neurol 2020;328:113233.
13. Santamaria-Cadavid M, Rodriguez-Castro E, Rodriguez-Yanez M, Arias-Rivas S, Lopez-Dequidt I, et al. Regulatory T cells participate in the recovery of ischemic stroke patients. BMC Neurol 2020;20:68.
14. Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med 2012;18:911-7.
15. Nishibori M, Mori S, Takahashi HK. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J Pharmacol Sci 2019;140:94-101.
16. Tuttolomondo A, Pecoraro R, Pinto A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: a review of the evidence to date. Drug Des Devel Ther 2014;8:2221-38.
18. Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, et al. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci 2001;21:5528-34.
19. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med 2009;15:946-50.
20. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 2015;6:7360.
21. Benakis C, Brea D, Caballero S, Faraco G, Moore J, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med 2016;22:516-23.
22. Gelderblom M, Gallizioli M, Ludewig P, Thom V, Arunachalam P, et al. IL-23 (Interleukin-23)-producing conventional dendritic cells control the detrimental IL-17 (Interleukin-17) response in stroke. Stroke 2018;49:155-64.
23. Mohammadi Shahrokhi V, Ravari A, Mirzaei T, Zare-Bidaki M, et al. IL-17A and IL-23: plausible risk factors to induce age-associated inflammation in Alzheimer’s disease. Immunol Invest 2018;47:812-22.
24. Waisman A, Hauptmann J, Regen T. The role of IL-17 in CNS diseases. Acta Neuropathol 2015;129:625-37.
25. Ren H, Kong Y, Liu Z, Zang D, Yang X, et al. Selective NLRP3 (Pyrin Domain-Containing Protein 3) inflammasome inhibitor reduces brain injury after intracerebral hemorrhage. Stroke 2018;49:184-92.
26. Cameron HA, Hazel TG, McKay RD. Regulation of neurogenesis by growth factors and neurotransmitters. J Neurobiol 1998;36:287-306.
27. Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, et al. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci 2015;9:147.
28. Locatelli G, Theodorou D, Kendirli A, Jordao MJC, Staszewski O, et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat Neurosci 2018;21:1196-208.
29. Shichita T, Ito M, Morita R, Komai K, Noguchi Y, et al. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat Med 2017;23:723-32.
30. Sakai S, Shichita T. Inflammation and neural repair after ischemic brain injury. Neurochem Int 2018;130:104316.
31. Tian X, An R, Luo Y, Li M, Xu L, et al. Tamibarotene improves hippocampus injury induced by focal cerebral ischemia-reperfusion via modulating PI3K/Akt pathway in rats. J Stroke Cerebrovasc Dis 2019;28:1832-40.
32. Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat Commun 2013;4:2030.
33. Cornejo F, von Bernhardi R. Role of scavenger receptors in glia-mediated neuroinflammatory response associated with Alzheimer’s disease. Mediators Inflamm 2013;2013:895651.
34. Giraldi-Guimaraes A, de Freitas HT, Coelho Bde P, Macedo-Ramos H, Mendez-Otero R, et al. Bone marrow mononuclear cells and mannose receptor expression in focal cortical ischemia. Brain Res 2012;1452:173-84.
35. Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 2016;7:11499.
36. Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 2003;278:43807-17.
37. Bazan NG, Eady TN, Khoutorova L, Atkins KD, Hong S, et al. Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp Neurol 2012;236:122-30.
38. Wu Y, Ye XH, Guo PP, Xu SP, Wang J, et al. Neuroprotective effect of lipoxin A4 methyl ester in a rat model of permanent focal cerebral ischemia. J Mol Neurosci 2010;42:226-34.
39. Sorce S, Bonnefont J, Julien S, Marq-Lin N, Rodriguez I, et al. Increased brain damage after ischaemic stroke in mice lacking the chemokine receptor CCR5. Br J Pharmacol 2010;160:311-21.
40. Joy MT, Ben Assayag E, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, et al. CCR5 is a therapeutic target for recovery after stroke and traumatic brain injury. Cell 2019;176:1143-57.e13.
41. Cowell RM, Xu H, Parent JM, Silverstein FS. Microglial expression of chemokine receptor CCR5 during rat forebrain development and after perinatal hypoxia-ischemia. J Neuroimmunol 2006;173:155-65.
42. Victoria ECG, de Brito Toscano EC, de Sousa Cardoso AC, da Silva DG, de Miranda AS, et al. Knockdown of C-C chemokine receptor 5 (CCR5) is protective against cerebral ischemia and reperfusion injury. Curr Neurovasc Res 2017;14:125-31.
43. Rawlinson C, Jenkins S, Thei L, Dallas ML, Chen R. Post-ischaemic immunological response in the brain: targeting microglia in ischaemic stroke therapy. Brain Sci 2020;10:159.
44. Tsuji S, Di Martino E, Mukai T, Tsuji S, Murakami T, et al. Aggravated brain injury after neonatal hypoxic ischemia in microglia-depleted mice. J Neuroinflammation 2020;17:111.
45. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005;8:752-8.
46. Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014;505:223-8.
47. Nonaka S, Nakanishi H. Microglial clearance of focal apoptotic synapses. Neurosci Lett 2019;707:134317.
48. Willis EF, MacDonald KPA, Nguyen QH, Garrido AL, Gillespie ER, et al. Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell 2020;180:833-46.e16.
49. De Geyter D, Stoop W, Sarre S, De Keyser J, Kooijman R. Neuroprotective efficacy of subcutaneous insulin-like growth factor-I administration in normotensive and hypertensive rats with an ischemic stroke. Neuroscience 2013;250:253-62.
50. Labandeira-Garcia JL, Costa-Besada MA, Labandeira CM, Villar-Cheda B, Rodriguez-Perez AI. Insulin-like growth factor-1 and neuroinflammation. Front Aging Neurosci 2017;9:365.
51. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 2010;87:779-89.
52. Doyle KP, Quach LN, Sole M, Axtell RC, Nguyen TV, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci 2015;35:2133-45.
53. Doyle KP, Buckwalter MS. Does B lymphocyte-mediated autoimmunity contribute to post-stroke dementia? Brain Behav Immun 2017;64:1-8.
54. Rojas OL, Probstel AK, Porfilio EA, Wang AA, Charabati M, et al. Recirculating intestinal IgA-producing cells regulate neuroinflammation via IL-10. Cell 2019;176:610-24.e18.
55. Planas AM, Gomez-Choco M, Urra X, Gorina R, Caballero M, et al. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol 2012;188:2156-63.
56. Cramer JV, Benakis C, Liesz A. T cells in the post-ischemic brain: troopers or paramedics? J Neuroimmunol 2019;326:33-7.
57. Clarkson BD, Ling C, Shi Y, Harris MG, Rayasam A, et al. T cell-derived interleukin (IL)-21 promotes brain injury following stroke in mice. J Exp Med 2014;211:595-604.
58. Lee HK, Keum S, Sheng H, Warner DS, Lo DC, et al. Natural allelic variation of the IL-21 receptor modulates ischemic stroke infarct volume. J Clin Invest 2016;126:2827-38.
59. Llovera G, Benakis C, Enzmann G, Cai R, Arzberger T, et al. The choroid plexus is a key cerebral invasion route for T cells after stroke. Acta Neuropathol 2017;134:851-68.
60. Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 2009;15:192-9.
61. Fu Y, Zhang N, Ren L, Yan Y, Sun N, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A 2014;111:18315-20.
62. Mracsko E, Liesz A, Stojanovic A, Lou WP, Osswald M, et al. Antigen dependently activated cluster of differentiation 8-positive T cells cause perforin-mediated neurotoxicity in experimental stroke. J Neurosci 2014;34:16784-95.
63. Gelderblom M, Arunachalam P, Magnus T. Gammadelta T cells as early sensors of tissue damage and mediators of secondary neurodegeneration. Front Cell Neurosci 2014;8:368.
64. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012;120:3793-802.
65. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab 2012;32:598-611.
66. Zhang H, Xia Y, Ye Q, Yu F, Zhu W, et al. In vivo expansion of regulatory T cells with IL-2/IL-2 antibody complex protects against transient ischemic stroke. J Neurosci 2018;38:10168-79.
67. Li P, Wang L, Zhou Y, Gan Y, Zhu W, et al. C-C chemokine receptor type 5 (CCR5)-mediated docking of transferred tregs protects against early blood-brain barrier disruption after stroke. J Am Heart Assoc 2017;6:e006387.
68. Liesz A, Kleinschnitz C. Regulatory T cells in post-stroke immune homeostasis. Transl Stroke Res 2016;7:313-21.
69. Xu X, Li M, Jiang Y. The paradox role of regulatory T cells in ischemic stroke. Scientific World Journal 2013;2013:174373.
70. Kipnis J, Schwartz M. Controlled autoimmunity in CNS maintenance and repair: naturally occurring CD4+CD25+ regulatory T-cells at the crossroads of health and disease. Neuromolecular Med 2005;7:197-206.
71. Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, et al. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nature immunology 2013;14:230-7.
72. McNeill A, Spittle E, Backstrom BT. Partial depletion of CD69low-expressing natural regulatory T cells with the anti-CD25 monoclonal antibody PC61. Scand J Immunol 2007;65:63-9.
73. Raposo C, Graubardt N, Cohen M, Eitan C, London A, et al. CNS repair requires both effector and regulatory T cells with distinct temporal and spatial profiles. J Neurosci 2014;34:10141-55.
74. Delacher M, Imbusch CD, Weichenhan D, Breiling A, Hotz-Wagenblatt A, et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat Immunol 2017;18:1160-72.
76. Sharma A, Rudra D. Emerging functions of regulatory T cells in tissue homeostasis. Front Immunol 2018;9:883.
77. Ito M, Komai K, Nakamura T, Srirat T, Yoshimura A. Tissue regulatory T cells and neural repair. Int Immunol 2019;31:361-9.
78. Wang K, Yaghi OK, Spallanzani RG, Chen X, Zemmour D, et al. Neuronal, stromal, and T-regulatory cell crosstalk in murine skeletal muscle. Proc Natl Acad Sci U S A 2020;10:5402-8.
79. DiSpirito JR, Zemmour D, Ramanan D, Cho J, Zilionis R, et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol 2018;3:eeat5861.
80. Hayatsu N, Miyao T, Tachibana M, Murakami R, Kimura A, et al. Analyses of a mutant Foxp3 allele reveal BATF as a critical transcription factor in the differentiation and accumulation of tissue regulatory T cells. Immunity 2017;47:268-83.e9.
81. Dombrowski Y, O’Hagan T, Dittmer M, Penalva R, Mayoral SR, et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci 2017;20:674-80.
82. Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 2015;85:703-9.
83. Guo S, Luo Y. Brain Foxp3(+) regulatory T cells can be expanded by Interleukin-33 in mouse ischemic stroke. Int Immunopharmacol 2019;81:106027.
84. Xiao W, Guo S, Chen L, Luo Y. The role of Interleukin-33 in the modulation of splenic T-cell immune responses after experimental ischemic stroke. J Neuroimmunol 2019;333:576970.
85. Li XM, Wang XY, Feng XW, Shao MM, Liu WF, et al. Serum interleukin-33 as a novel marker for long-term prognosis and recurrence in acute ischemic stroke patients. Brain Behav 2019;9:e01369.
86. Lenglet S, Louiset E, Delarue C, Vaudry H, Contesse V. Activation of 5-HT(7) receptor in rat glomerulosa cells is associated with an increase in adenylyl cyclase activity and calcium influx through T-type calcium channels. Endocrinology 2002;143:1748-60.
87. Klein M, Bopp T. Cyclic AMP represents a crucial component of Treg cell-mediated immune regulation. Front Immunol 2016;7:315.
88. Sacramento PM, Monteiro C, Dias ASO, Kasahara TM, Ferreira TB, et al. Serotonin decreases the production of Th1/Th17 cytokines and elevates the frequency of regulatory CD4(+) T-cell subsets in multiple sclerosis patients. Eur J Immunol 2018;48:1376-88.
89. Gu SC, Wang CD. Early selective serotonin reuptake inhibitors for recovery after stroke: a meta-analysis and trial sequential analysis. J Stroke Cerebrovasc Dis 2018;27:1178-89.
90. Chollet F, Tardy J, Albucher JF, Thalamas C, Berard E, et al. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol 2011;10:123-30.
91. FOCUS-Trial-Collaboration. Effects of fluoxetine on functional outcomes after acute stroke (FOCUS): a pragmatic, double-blind, randomised, controlled trial. Lancet 2019;393:265-74.
92. Panickar KS, Norenberg MD. Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia 2005;50:287-98.
93. Xu Y, Meng C, Liu G, Yang D, Fu L, et al. Classically activated macrophages protect against lipopolysaccharide-induced acute lung injury by expressing amphiregulin in mice. Anesthesiology 2016;124:1086-99.
94. Abeysinghe HC, Phillips EL, Chin-Cheng H, Beart PM, Roulston CL. Modulating astrocyte transition after stroke to promote brain rescue and functional recovery: emerging targets include rho kinase. Int J Mol Sci 2016;17:288.
95. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541:481-7.
96. Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 2006;12:829-34.
97. Kimura H, Schubert D. Schwannoma-derived growth factor promotes the neuronal differentiation and survival of PC12 cells. J Cell Biol 1992;116:777-83.
98. Zelenika D, Adams E, Humm S, Graca L, Thompson S, et al. Regulatory T cells overexpress a subset of Th2 gene transcripts. J Immunol 2002;168:1069-79.
99. Liska MG, Crowley MG, Tuazon JP, Borlongan CV. Neuroprotective and neuroregenerative potential of pharmacologically-induced hypothermia with D-alanine D-leucine enkephalin in brain injury. Neural Regen Res 2018;13:2029-37.
100. Zagon IS, Rahn KA, Bonneau RH, Turel AP, McLaughlin PJ. Opioid growth factor suppresses expression of experimental autoimmune encephalomyelitis. Brain Res 2010;1310:154-61.
101. Weir C, McNeill A, Hook S, Harvie M, La Flamme AC, et al. Critical role of preproenkephalin in experimental autoimmune encephalomyelitis. J Neuroimmunol 2006;179:18-25.
102. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012;486:549-53.
103. Villapol S. Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol Neurobiol 2018;38:121-32.
104. Yasuno F, Taguchi A, Yamamoto A, Kajimoto K, Kazui H, et al. Microstructural abnormality in white matter, regulatory T lymphocytes, and depressive symptoms after stroke. Psychogeriatrics 2014;14:213-21.
105. Swardfager W, Herrmann N, Andreazza AC, Swartz RH, Khan MM, et al. Poststroke neuropsychiatric symptoms: relationships with IL-17 and oxidative stress. Biomed Res Int 2014;2014:245210.
106. Dolati S, Ahmadi M, Khalili M, Taheraghdam AA, Siahmansouri H, et al. Peripheral Th17/Treg imbalance in elderly patients with ischemic stroke. Neurol Sci 2018;39:647-54.
107. Yan J, Read SJ, Henderson RD, Hull R, O’Sullivan JD, et al. Frequency and function of regulatory T cells after ischaemic stroke in humans. J Neuroimmunol 2012;243:89-94.
108. Pang X, Qian W. Changes in regulatory T-cell levels in acute cerebral ischemia. J Neurol Surg A Cent Eur Neurosurg 2017;78:374-9.
109. Duffy SS, Keating BA, Perera CJ, Moalem-Taylor G. The role of regulatory T cells in nervous system pathologies. J Neurosci Res 2018;96:951-68.
110. Lunn JS, Sakowski SA, McGinley LM, Pacut C, Hazel TG, et al. Autocrine production of IGF-I increases stem cell-mediated neuroprotection. Stem Cells 2015;33:1480-9.
111. Falk A, Frisen J. Amphiregulin is a mitogen for adult neural stem cells. J Neurosci Res 2002;69:757-62.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yoshimura A, Ito M. Resolution of inflammation and repair after ischemic brain injury. Neurosciences 2020;7:264-76. http://dx.doi.org/10.20517/2347-8659.2020.22
AMA Style
Yoshimura A, Ito M. Resolution of inflammation and repair after ischemic brain injury. Neuroimmunology and Neuroinflammation. 2020; 7(3): 264-76. http://dx.doi.org/10.20517/2347-8659.2020.22
Chicago/Turabian Style
Yoshimura, Akihiko, Minako Ito. 2020. "Resolution of inflammation and repair after ischemic brain injury" Neuroimmunology and Neuroinflammation. 7, no.3: 264-76. http://dx.doi.org/10.20517/2347-8659.2020.22
ACS Style
Yoshimura, A.; Ito M. Resolution of inflammation and repair after ischemic brain injury. Neurosciences. 2020, 7, 264-76. http://dx.doi.org/10.20517/2347-8659.2020.22
About This Article
Copyright

Data & Comments
Data


 Cite This Article 41 clicks
Cite This Article 41 clicks

 Like This Article 18
likes
Like This Article 18
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.