
In silico design of novel gold-phosphate containing compounds as selective inhibitors of cathepsin B in neuroinflammation
 0
0Abstract
Aim: Alzheimer’s disease is characterized by pathological protein aggregates and microglia-driven chronic neuroinflammation. Cathepsin B has been proposed as the potential target for inhibiting adverse activation of microglia and slowing down this neurodegenerative disease. Currently available inhibitors of cathepsin B enzymatic activity are non-selective; therefore, the design and synthesis of novel specific inhibitors could facilitate the development of a new class of anti-Alzheimer medications targeting the neuroinflammatory component of this disease.
Methods: We describe molecular design strategies, which were used to create specific cathepsin B inhibitors based on the structure of the gold-containing drug auranofin (Ridaura), and its covalent binding to the cysteine residue of the active site of cathepsins.
Results: This in silico study investigated the structure-activity relationship of a series of newly designed derivatives of auranofin with regard to their cathepsin B inhibitory activity. An exhaustive molecular screening model was designed and validated by using a set of known cathepsin B inhibitors. Its validity was further tested during the preliminary stage of the biological screening of newly designed inhibitors. Based on the structure-function relationships discovered by recording the empirical score values generated for the screening model, a series of subsequent in silico predictions of compound inhibitory activity were generated, which led to new structures with increased inhibitory activity and selectivity towards cathepsin B.
Conclusion: The described molecular modeling strategy could be employed to design novel inhibitors of cathepsin B enzymatic activity, which could be used to slow down neuroinflammation in neurodegenerative disorders including Alzheimer’s disease.
Keywords
Introduction
Alzheimer’s disease is the most common cause of dementia. It is characterized by abnormal brain protein deposits including amyloid ß (Aß)-containing plaques and neurofibrillary tangles. In addition, chronic neuroinflammation, driven by adverse activation of non-neuronal microglial cells, is believed to contribute to the pathogenesis of this neurodegenerative disease. Microglial immune functions can be regulated by various cathepsin enzymes, many of which are the components of lysosomes[1,2]. Cathepsins belong to the papain superfamily and are synthesized as inactive pro-enzymes[3]. Cathepsin B, a cysteine protease, is expressed and can be secreted by activated microglia[4-7]. Cathepsin B has been proposed as a potential therapeutic target to reduce neuroinflammation in Alzheimer’s disease[8] based on observations showing this protein upregulated in brain tissues, serum and cerebrospinal fluid of Alzheimer’s patients[9-13]. Such high levels of cathepsin B also correlate with decreased cognition, which could be caused by adverse microglia activation[11,14].
A series of pre-clinical studies support using cathepsin B inhibitors to slow the progression of Alzheimer’s disease. Reduced Aß plaque load and improved memory were reported in a transgenic Alzheimer mice model after deleting the cathepsin B gene[15]. In vitro studies showed that cathepsin B inhibitors downregulated pro-inflammatory cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-1ß, and IL-18[16-18]. Cathepsin B has been linked to specific microglial functions including their secretion of cytokines, neurotoxicity, and Aß degradation[5,17,19]. Currently available cathepsin B inhibitors are not specific since they inhibit enzymatic activity of other cysteine proteases including calpains, as well as cathepsins S and L[3,20-23]. Therefore, novel highly selective cathepsin B inhibitors should be developed and tested for their ability to ameliorate neuroinflammation in neurodegenerative diseases.
Cathepsin B enzymatic activity is inhibited by alpha-macroglobulin from the cystatin family of inhibitors of papain-like cysteine peptidases, and by representatives of the equistatin family[24]. There are three additional groups of naturally occurring cathepsin B inhibitors: the aziridinyl peptides, peptide epoxysuccinyls, and peptide aldehydes[25,26]. Known synthetic cathepsin B inhibitors can be divided into groups of compounds, which contain either flavonoids, cyclic sulfates, or nitriles[27,28]. Cathepsins B and K are inhibited with reasonable potency by gold(I)-based compounds such as auranofin (Ridaura), which is clinically used as an anti-rheumatic agent, and its analogs[29]. Structure-activity relationship (SAR) studies revealed that replacement of ethyl substituent with a voluminous aryl substituent in auranofin, which is a clinically approved triethylphosphine (PEt3) gold-containing drug, significantly increased its anti-inflammatory activity[30]. A triphenylphosphine gold compound was shown to be a more effective cathepsin B inhibitor (IC50 = 337 nmol/L) than its parent compound auranofin (IC50 > 250 µmol/L)[30]. Further studies confirmed that compounds with more than one aryl group (e.g., triarylphosphines) were much stronger cathepsin B inhibitors than triethylphosphine Au(I)-containing derivatives[31].
In this study, we describe an in silico model developed for identification of pharmacophores capable of increasing the biological activity of Au(I)-based drug-like compounds. Previously, it was shown that changes in steric and electronic properties of phosphine derivatives led to increased affinity of these compounds toward cathepsin B[31]. The aim of the present study was to develop and test novel structural modifications of cathepsin B inhibitors, which could be used for synthesis of new, potentially more effective anti-neuroinflammatory drugs. To achieve this goal, we first developed an in silico docking model of the cathepsin B enzymatic pocket. Subsequently, a series of novel cathepsin B inhibitors were designed and synthesized, based on their calculated binding affinity to the enzymatic pocket and docking scores. An in vitro testing of selected compounds as possible cathepsin B inhibitors was also performed.
Methods
In silico modeling
All manipulations with protein-ligand structures and generation of structural models of cathepsin-ligand complexes were performed with Sybyl-X software (Tripos Inc., St. Louis, MO, USA). The three-dimensional structure of the triethylphosphine was generated by the CONCORD version 3.0 software (CONCORD, St Louis, MO, USA). Ligand topologies for molecular dynamics (MD) studies were calculated using the antechamber module of AmberTools version 12[32]. The protein structures of cathepsins B and K (Protein Data Bank Identifiers (PDB IDs): 1HUC and 2ATO) were obtained from the Research Collaboratory for Structural Bioinformatics (RCSB, www.rcsb.org)[33]. Structures of the complexes formed by cathepsin B interacting with triethylphosphine, as well as cathepsin K with myochrysine (Au-thiomalate), were modeled in the experiment. The subsequent steps of the relaxation strategy for the structural optimization of active site geometry were carried out with Gromacs (version 4.5) software with implementation of Amber99 force field[34].
Due to the absence of defined atom-type and coordination bonding parameters for Au in both MD and docking software, we utilized fragment-based docking methods for the model development. Replacement of the Au atom with a CF3 group, which has four heavy atoms, allowed us to preserve both geometry and distances between the ligand and C-alpha of the catalytic residue Cys29 (C29) in the cathepsin B molecule [Figure 1]. As a result of this manipulation, a “joint-like” metal-based binding of Au was replaced with the “anchor-like” binding mode of CF3. Additional mutation of the catalytic cysteine (C29) to alanine (C29A) provided more flexibility for the CF3-containing ligands and avoided artificial interaction between the sulfur atom of C29 and the CF3 group.
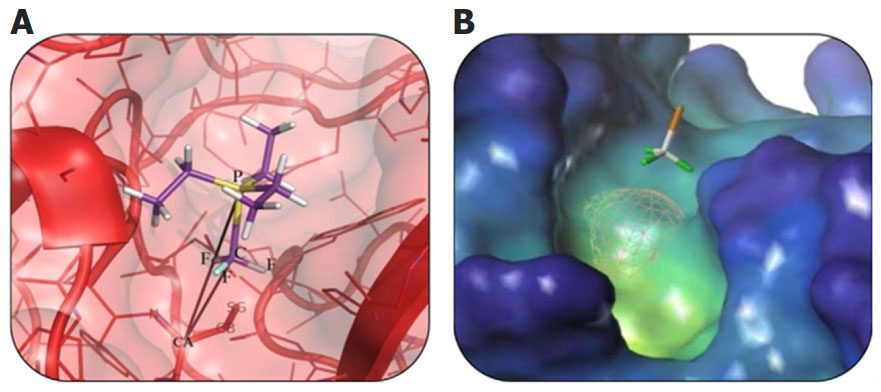
Figure 1. A schematic representation of the mutual orientation and disposition of P-CF3 part of triethylphosphine and C-alpha (CA) atom of the Cys29 residue of cathepsin B (A). Active site of cathepsin В with the CF3 group (F atoms are colored green) (B). Cys29 is replaced with Ala29 (the surface of Cys29 is shown in mesh)
In order to remove any unfavorable steric clashes between the proteases and the inhibitor molecules, 1000 steps of steepest descent and 5000 steps of conjugate-gradient energy minimization were employed. The complex between cathepsin B and triethylphosphine was solvated with a 12 Å radius water box centered at the C29A. A center of mass (COM) pulling mode was applied during all steps of MD calculations to improve the geometry of the P-CF3-C-alpha (CA, of alanine) bridge. The optimized and equilibrated system was used as the starting configuration for MD simulations spanning 500 ps. Such fast molecular dynamics was enough to stabilize the group of constrained P-CF3-CA atoms. Average root-mean-square deviation (RMSD) values for the whole structure, including the active site and ligand atoms, were calculated from each of the five picoseconds frames of the simulation. RMSD values, together with the structure alignment, confirmed that conformation of the ligand and the active site of protein had not changed dramatically and were considered stable enough to continue modeling.
For the docking procedure, a specific binding site representation (namely Protomol) was generated based on the relaxed protein-ligand complex. Protomol is an interaction map, implemented in the Surflex-Dock module, which is based on the probing method used[35]. Protomol model is very sensitive to input parameters; therefore, to avoid mistakes in predictions, it was parameterized based on scores obtained through validation docking procedures. To keep the amino acid environment flexible during each docking run, the most reliable positions of the reference PEt3 were retained with the number of starting conformations per ligand higher than ten. The new compounds were designed based on the shape of the binding pocket and its hydrophobic/hydrophilic areas.
A set of reference compounds was compiled from publicly available structures and contained the triethylphosphine molecule and its modifications. Structures of these reference compounds and their corresponding IC50 values as cathepsin B inhibitors have been previously published[36]. Predictions from the docking studies were made based on correlations between docking scores and known enzymatic activity of the reference compounds. Surflex-Dock scores (total score expressed in: -log10 (Kd) units) represented the binding affinities. Four different calculated parameters were used as the most important indicators in docking analysis: “total score”, “crash value” (the degree of inappropriate penetration by the ligand into the protein; values close to zero are favorable), “internal ligand strain” and “complex absolute energy”. In addition, the root-mean-square (RMS) distance between the docked ligand and the reported fragment RMSD (FragRMSD) was calculated to provide the correct orientation of the ligand in the binding site.
In vitro testing
Cathepsin B enzymatic activity was measured as described by Hulkower et al.[37] (2000) with modifications. A fluorometric assay was used in which cathepsin B (from bovine spleen) cleaved its substrate, Z-Arg-Arg-AMC (both from Sigma Aldrich, Oakville, ON, Canada), causing the cleaved product to fluoresce. The reaction was performed in Hanks’ balanced salt solution containing 0.6 mmol/L CaCl2, 0.6 mmol/L MgCl2, 2 mmol/L L-cysteine, and 25 mmol/L PIPES, pH 7.0. The assay was performed in a 96-well plate, and the POLARstar Omega plate reader (BMG Labtech, Durham, NC, USA) with an excitation wavelength of 355 nm and an emission wavelength of 460 nm was used to measure the velocity of the reaction (relative fluorescence units per min) at 37 °C. Au-containing substances were dissolved in dimethyl sulfoxide (DMSO) and added to the reaction mixture at 10 nM to 500 μmol/L concentration range. The final concentration of the solvent in the reaction mixture did not exceed 0.5%. The solutions containing cathepsin B (200 μg/mL) and the inhibitors were allowed to incubate for 30 min at 37 °C. Subsequently Z-Arg-Arg-AMC (from a 600 μmol/L stock solution) was added to the wells to reach the final concentration of 30 μmol/L in a total well volume of 180 μL. The 96-well plate was then positioned into the plate reader and fluorescence measurements from each well were recorded. The plate reader was set to acquire 64 measurements over a 52-min time frame. The maximum slope values of the samples containing inhibitors were calculated as percentages of maximum slope values of the control samples containing DMSO vehicle solution only, and IC50 values for each inhibitor determined.
Results
Proper selections of binding sites, together with a good understanding of the mechanism of enzymatic activity of cathepsins, are the key factors for designing effective new inhibitors; however, currently available inhibitors of cathepsin B are only 2-to 8-fold more selective towards this enzyme compared to two other cathepsins S or L[3,22]. It was previously shown that cathepsin B is reversibly and competitively inhibited by linear Au(I)-containing complexes[29]. A hit-to-lead optimization of the organic ligands used to create new auranofin analogs, particularly the phosphine ligands, could significantly increase the potency of these compounds as cathepsin B inhibitors. Phosphine ligand interaction with cathepsin B was modeled based on the myochrysine (Au-thiomalate) binding to cathepsin K[31]. The crystal structure of the 339 amino acid-long human liver cathepsin B has been published at a 1.9 Å resolution (PDB ID: 1GYM)[38].
Access to the active site of cathepsin B is provided by an 18 amino acid long insertion (Pro107Asp124), termed the occluding loop, which possesses two His residues for binding of the carboxyl group of the substrate. Three-dimensional alignment of the crystal structures of cathepsins B, S, K and L demonstrated a considerable degree of homology, especially in the region of substrate binding. Our molecular modeling identified that the potential binding site on the surface of cathepsins is represented by a long hydrophobic pocket, which is necessary for the peptide binding and excision. Figure 2 shows that the structures of cathepsins K and B are very similar, including their cleavage sites containing several “hot-spot” amino acids conserved among all types of cathepsins, along with Cys29, which forms a coordination bond with PEt3.
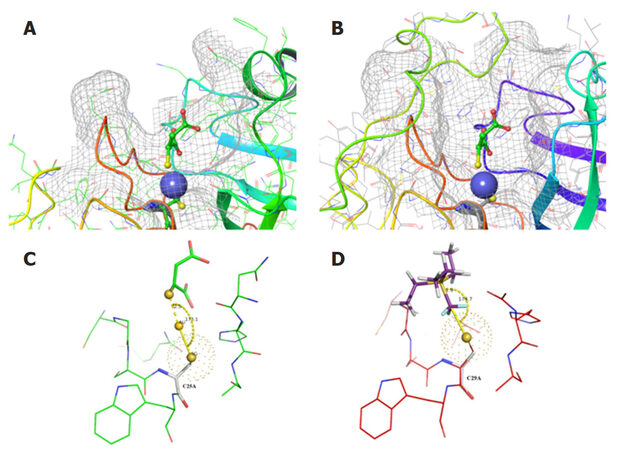
Figure 2. Cathepsin K (A) with co-crystalized and cathepsin B (B) with superimposed myochrysine (Au-thiomalate) are represented in mesh surface and colored ribbon style. Au atom is shown as a blue sphere. A secondary structure color scheme (suggests different colors for different secondary structures) was used for ribbon representation. A schematic representation of ligand binding mode and the geometry of the Au atom and the sulfur bridge in cathepsins K (C) and B (D). Ionic bond length and angle between the terminal sulfur of Cys25 of cathepsin K are indicated. The same view of CF3-substituted triethylphosphine in the active site of cathepsin B with mutated Cys29 to Ala29 is shown in (D)
Despite the great variety of protein modeling tools and techniques available, there is no simple approach to study interactions between Au-containing compounds and proteins without the time-consuming quantum mechanics (QM) calculations. The absence of Au or any cognate element and its bond parameters in most force field and docking protocols compelled us to implement several alternative techniques, known as steered molecular dynamics and fragment-based docking approach, as suitable models for docking and subsequent lead optimization. A structure preparation tool from SybylX suite was used to model the complex between cathepsin B and Au-containing ligand. A crystal structure of cathepsin B (PDB ID: 1HUC) was superimposed on the cathepsin K structure (PDB ID:2ATO) and coordinates of co-crystalized myochrysine (Au-thiomalate) and cathepsin B were saved for subsequent analyses. The highly conserved cysteine, histidine and asparagine residues from the active site of cathepsins B and K were used for structure superposition.
The measured essentially important linear angle between the atoms in the S-Au-S triad (sulfur of C25, Au(I) and sulfur of thiomalate) of the complex between cathepsin K and Au-thiomalate was 173.1o. Subsequently, the gold atom in Au-thiomalate was replaced with a CF3 group. CF3 is a directional and rigid group, which has a binding mode similar to the gold atom. The CF3 possesses four tetrahedral heavy atoms, which are necessary for accurate geometric constraints as well as the bond-like rotation during molecular dynamics and docking. In addition, the Cys25 residue in the enzymatic pocket of cathepsins B and K was replaced with alanine to eliminate a steric clash and electrostatic interactions with the CF3 group of thiomalate. In order to validate the docking procedure, a model of cathepsin K was developed based on its complex with CF3-containing Au-thiomalate. Subsequently, CF3-containing Au-thiomalate in this complex was replaced with CF3-substituted triethylphosphine.
The fast molecular dynamics simulation, spanning 500 ps, generated parameters of trajectories, which were analyzed and clustered by RMSD to identify the most stable conformation of the complexes. The docking model of cathepsin B was developed based on this structure. Figure 3 shows the resulting modified compound CF3-PEt3 positioned in the mutated (C29A) enzymatic pocket of cathepsin B, with preserved original geometry (position and orientation) of the PEt3 group. This structure maintained all the distances and co-linear characteristics of the complex between CF3-substituted triethylphosphine and cathepsin B. The volume of the cathepsin B active site was defined with a Protomol generation tool.
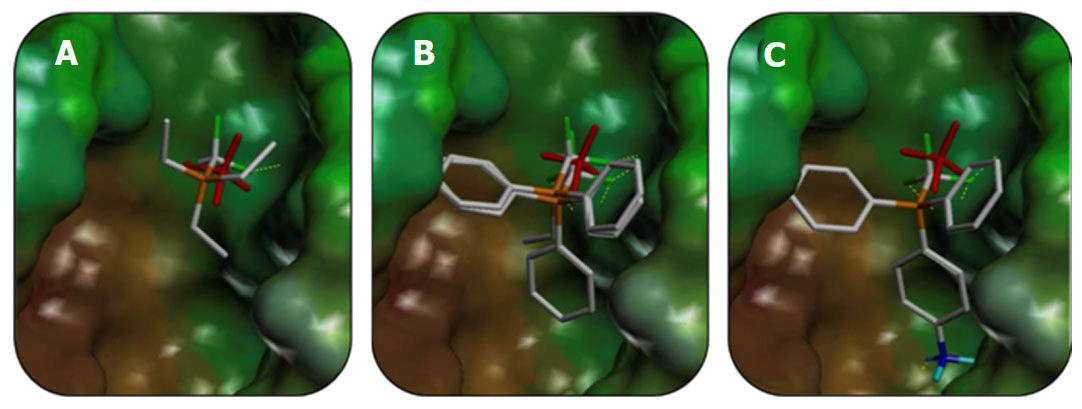
Figure 3. Orientations of the most potent reference enzymatic activity inhibitors P(CH2CH3)3 (A), P(CH2CH3)(C6H5)2 (B), and P(C6H5)3, P(C6H4NH3+)(C6H5)2 (C) in the active site of cathepsin B
Next, to provide ligand dislocation similar to that in the CF3-substituted complex between triethylphosphine and cathepsin B, we applied both the distance and position constraints. The Patom-CF3 fragment was assigned as a constraint in Surflex-Dock, to match the correct position and orientation of the ligands in the enzymatic pocket. After several test dockings we chose an appropriate value for “cpen” function (penalty for deviating from fragment), which determines how closely compounds could be positioned to the source coordinates of the template P-CF3 group of six different CF3-substituted auranofin derivatives. Reference compounds from an open source database and previous publications were selected for validation of the MD model[36,39]. Through the design of novel compounds, we aimed to improve their cathepsin B binding affinities. The three-dimensional structure of the new compounds was generated by the CONCORD (see Methods section). We chose a triphenylphosphine (PPh3) as the initial structure for design, since previous studies[29] confirmed its significant in vitro activity as an inhibitor of cathepsin B enzymatic activity. The validation step of the fragment-based docking defined optimal software settings, which provided the best correlation between the enzymatic activity inhibition data and the docking score [Table 1].
Scoring values of ligands docked in the active site of cathepsin B
| Sample | Total score | Crash score | Internal strain₸ | Complex energy | FragRMSD# | IC50 (µmol/L) |
|---|---|---|---|---|---|---|
| CF3P(CH2CH3)3 Triethylphosphine | 1.325 | -0.211 | 0.073 | 482.7 | 0.745 | ~250 |
| CF3P(C6H5)2(CH2C6H5) | 3.602 | -0.600 | 0.252 | 626.0 | 0.970 | ~64 |
| CF3P(CH2CH3)(C6H5)2 | 4.051 | -0.952 | 5.485 | 560.0 | 0.737 | 18.0 |
| *Cmpd 4 | 4.201 | -1.505 | 2.500 | 658.5 | 0.97 | 8.67 |
| Cmpd 2 | 3.991 | -0.003 | 2.449 | 594.1 | 1.123 | 1.00 |
| Cmpd 5 | 6.560 | -1.080 | 2.201 | 660.0 | 0.85 | 0.71 |
| Cmpd 3 | 4.059 | -0.919 | 0.918 | 624.4 | 0.997 | 0.46 |
| Cmpd 1 | 4.747 | -0.937 | 0.157 | 575.0 | 0.617 | 0.34 |
| CF3P(C6H5)3 | 4.353 | -1.150 | 2.001 | 607.6 | 0.840 | 0.33 |
| CF3P(C6H4-C6H5)(C6H5)2 | 4.401 | -1.301 | 0.452 | 613.1 | 0.861 | 0.29 |
| CF3P(C6H4NH3+)(C6H5)2 | 6.940 | -1.130 | 3.157 | 731.2 | 1.002 | 0.20 |
| Cmpd 6 | 6.938 | -1.640 | 0.4503 | 703.6 | 0.80 | 0.17 |
Table 1 lists a set of compounds with known IC50 values in cathepsin B enzymatic assays. This table also includes the previously reported compound triethylphosphine (P(CH2CH3)3) and its analogs with substituted methyl groups on phenyl rings[31]. Table 1 likewise includes examples of newly synthesized compounds (Cmpd 1-6). These novel derivatives were created by replacing the triethylphosphine ligand of the auranofin molecule with other phosphines carrying various aromatic and heteroaromatic substituents. Table 1 is divided into seven columns and the most practical parameter for the docking analysis outcome is the summarized score (total score), which is an empirical affinity value developed to predict the potency of the inhibitors. This value is calculated based on the other parameters shown in this table. The upper part of the table illustrates correlations between scores obtained from in silico modeling and in vitro testing described in published studies and patents[31,36,40]. The lower part of the table shows predicted affinities and the measured cathepsin B inhibitor activities for the newly synthesized compounds. The validation step was complete after confirmation of a relationship between the calculated and biochemically determined values for known inhibitors. These two parameters are listed in Table 1 and compounds are sorted in order of ascending IC50 values in cathepsin B enzymatic activity assay, which correlate inversely (negative correlation coefficient of -0.71) with the calculated total score values.
In addition, we repeated all docking procedures with cathepsin K, exactly as they were described for cathepsin B above, by applying match constraining to CF3. In this way, we compared the results of our MD modeling for both proteins, which showed different docking scores for the interactions between the substituted triethylphosphine derivatives and cathepsin K [Table 2]. These data indicate a certain degree of selectivity towards a specific cathepsin isoform for these compounds.
Scoring values of ligands docked in the active site of cathepsin K
| Sample | Total score | Crash score | Internal strain | Complex energy | Fragment RMSD* |
|---|---|---|---|---|---|
| CF3P(CH2CH3)3 triethylphosphine | 1.3065 | -0.4537 | 0.0682 | 369.7 | 0.694 |
| CF3 Au-thiomalate | 1.984 | -0.3104 | 0.034 | 317.7 | 0.705 |
| CF3P(CH2CH3)(C6H5)2 | 2.4274 | -0.4531 | 0.0663 | 390.3 | 0.528 |
| CF3P(C6H5)3 | 3.4247 | -1.4016 | 4.5 | 451.1 | 0.901 |
Three-dimensional alignments of docked reference inhibitors in the active site of cathepsin B are presented in Figure 3. RMSD values for the compounds used to estimate the three-dimensional matching of the reference arylphosphine derivatives to substituted triethylphosphine are listed in Table 1. After the initial lead compound optimization and docking, a limited set of seven substituted phosphines were synthesized from a total of 15 designed structures and used for in vitro studies. The best way of comparing the overall calculated binding affinities of ligands is a comparison of their total score values. However, all other scoring functions can be useful in specific cases. For example, as can be seen from Table 1, the best experimental and docking results were obtained for P(C6H4NH3+)(C6H5)2 compound. It is evident that high total scores and low internal strain scores are preferable for the newly developed compounds. Therefore, these parameters were used for the further design of the improved novel drug-like compounds. Further optimization and improvement of each drug-like compound were based on their spatial location in the enzymatic pocket of cathepsin B and compliance to the physical features of the active site, including anchoring with the CF3 group and localization of phosphine groups in the hydrophobic/hydrophilic regions of the enzymatic pocket.
Discussion
Our data show that the described docking model is a practical tool for identification and optimization of novel compounds, which have been designed based on their phosphine core structure. We also focused our attention on the shape features of the cathepsin B binding site and achieved a good trend in selectivity of the inhibitors towards this enzyme, by avoiding their binding to cathepsin K. The inverse correlation (-0.71) between docking scores of compounds and their IC50 values as well as significant selectivity towards cathepsin B and not cathepsin K [Tables 1 and 2] supported subsequent docking analysis and selection of compounds for in vitro testing. Thus, by using the molecular modeling described in this study we were able to design and create structurally novel derivatives of the clinically available anti-rheumatic drug auranofin, which inhibited the enzymatic activity of cathepsin B more effectively than their parent drug. It has been established that the clinical anti-inflammatory activity of auranofin depends on its ability to affect multiple cellular and molecular targets[41]. Even though clinical effectiveness of auranofin in neurodegenerative disorders has not been studied, its anti-inflammatory activity may be beneficial for slowing down neuroinflammation accompanying such pathologies as Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis[42]. Optimizing the activity and selectivity of auranofin molecule as an inhibitor of cathepsin B through structural modifications may lead to additional benefits of such novel compounds in Alzheimer’s disease in particular[43]. Further in vivo studies will be required to determine the pharmacokinetics and pharmacodynamics of these novel derivatives of auranofin, as well as their clinical suitability as anti-neuroinflammatory drugs and cathepsin B inhibitors.
Declarations
Authors’ contributionsConceived the study and wrote the manuscript: Raevsky AV, Sharifi M, Pinchuk V, Klegeris A
Conducted modeling experiments and analyzed the data: Raevsky AV, Sharifi M
Availability of data and materialsData in this study were obtained by experimentation and through in silico modeling, and are original. All primary data used to construct the figures and summary tables are available by contacting the authors of this study.
Financial support and sponsorshipThis work was supported by a grant from the Jack Brown and Family Alzheimer’s Disease Research Foundation. The authors acknowledge support for this study from Life Chemicals Ltd. (Kiev, Ukraine; http://www.lifechemicals.com).
Conflicts of interestThe authors declare no conflict of interest. M. Sharifi declares the view presented in this article are those of the author and do not reflect those of the US Food and Drug Administration. No official endorsement is intended nor should be inferred.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
2. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 2012;1824:68-88.
3. Siklos M, BenAissa M, Thatcher GR. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B 2015;5:506-19.
4. Gan L, Ye S, Chu A, Anton K, Yi S, Vincent VA, von Schack D, Chin D, Murray J, Lohr S, Patthy L, Gonzalez-Zulueta M, Nikolich K, Urfer R. Identification of cathepsin B as a mediator of neuronal death induced by A beta-activated microglial cells using a functional genomics approach. J Biol Chem 2004;279:5565-72.
5. Kingham PJ, Pocock JM. Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem 2001;76:1475-84.
6. Liuzzo JP, Petanceska SS, Devi LA. Neurotrophic factors regulate cathepsin S in macrophages and microglia: A role in the degradation of myelin basic protein and amyloid beta peptide. Mol Med 1999;5:334-43.
7. Ryan RE, Sloane BF, Sameni M, Wood PL. Microglial cathepsin B: an immunological examination of cellular and secreted species. J Neurochem 1995;65:1035-45.
8. Hook G, Jacobsen JS, Grabstein K, Kindy M, Hook V. Cathepsin B is a new drug target for traumatic brain injury therapeutics: evidence for E64d as a promising lead drug candidate. Front Neurol 2015;6:178.
9. Cataldo AM, Nixon RA. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc Natl Acad Sci U S A 1990;87:3861-5.
10. Nakamura Y, Takeda M, Suzuki H, Hattori H, Tada K, Hariguchi S, Hashimoto S, Nishimura T. Abnormal distribution of cathepsins in the brain of patients with Alzheimers disease. Neurosci Lett 1991;130:195-8.
11. Sun Y, Rong X, Lu W, Peng Y, Li J, Xu S, Wang L, Wang X. Translational study of Alzheimer's disease (AD) biomarkers from brain tissues in AβPP/PS1 mice and serum of AD patients. J Alzheimers Dis 2015;45:269-82.
12. Sundelöf J, Sundström J, Hansson O, Eriksdotter-Jönhagen M, Giedraitis V, Larsson A, Degerman-Gunnarsson M, Ingelsson M, Minthon L, Blennow K, Kilander L, Basun H, Lannfelt L. Higher cathepsin B levels in plasma in Alzheimer’s disease compared to healthy controls. J Alzheimers Dis 2010;22:1223-30.
13. Zhang J, Goodlett DR, Quinn JF, Peskind E, Kaye JA, Zhou Y, Pan C, Yi E, Eng J, Wang Q, Aebersold RH, Montine TJ. Quantitative proteomics of cerebrospinal fluid from patients with Alzheimer disease. J Alzheimers Dis 2005;7:125-33.
14. Ni J, Wu Z, Peterts C, Yamamoto K, Qing H, Nakanishi H. The critical role of proteolytic relay through cathepsins B and E in the phenotypic change of microglia/macrophage. J Neurosci 2015;35:12488-501.
15. Kindy MS, Yu J, Zhu H, El-Amouri SS, Hook V, Hook GR. Deletion of the cathepsin B gene improves memory deficits in a transgenic Alzheimer's disease mouse model expressing AβPP containing the wild-type β-secretase site sequence. J Alzheimers Dis 2012;29:827-40.
16. Ha SD, Martins A, Khazaie K, Han J, Chan BM, Kim SO. Cathepsin B is involved in the trafficking of TNF-alpha-containing vesicles to the plasma membrane in macrophages. J Immunol 2008;181:690-7.
17. Terada K, Yamada J, Hayashi Y, Wu Z, Uchiyama Y, Peters C, Nakanishi H. Involvement of cathepsin B in the processing and secretion of interleukin-1beta in chromogranin A-stimulated microglia. Glia 2010;58:114-24.
18. Wendt W, Schulten R, Stichel CC, Lübbert H. Intra- versus extracellular effects of microglia-derived cysteine proteases in a conditioned medium transfer model. J Neurochem 2009;110:1931-41.
19. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008;9:857-65.
20. Huang Z, Mcgowan EB, Detwiler TC. Ester and amide derivatives of E64c as inhibitors of platelet calpains. J Med Chem 1992;35:2048-54.
21. Katunuma N. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc Jpn Acad Ser B Phys Biol Sci 2011;87:29-39.
22. Smith HJ, Simons C. Protease and peptidase inhibition: recent potential targets for drug development. London: CRC Press; 2002. pp. 94-144.
23. Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes 2004;53 Suppl 1:S12-8.
25. Ito H, Watanabe M, Kim YT, Takahashi K. Inhibition of rat liver cathepsins B and L by the peptide aldehyde benzyloxycarbonyl-leucyl-leucyl-leucinal and its analogues. J Enzyme Inhib Med Chem 2009;24:279-86.
26. Schaschke N, Deluca D, Assfalg-Machleidt I, Hohneke C, Sommerhoff CP, Machleidt W. Epoxysuccinyl peptide-derived cathepsin B inhibitors: Modulating membrane permeability by conjugation with the C-terminal heptapeptide segment of penetratin. Biol Chem 2002;383:849-52.
27. Quesne MG, Ward RA, de Visser SP. Cysteine protease inhibition by nitrile-based inhibitors: a computational study. Front Chem 2013;1:39.
28. Ramalho SD, de Sousa LR, Burger MC, Lima MI, da Silva MF, Fernandes JB, Vieira PC. Evaluation of flavonols and derivatives as human cathepsin B inhibitor. Nat Prod Res 2015;29:2212-4.
29. Gunatilleke SS, Barrios AM. Inhibition of lysosomal cysteine proteases by a series of Au(I) complexes: a detailed mechanistic investigation. J Med Chem 2006;49:3933-7.
30. Berners-Price SJ. Gold-based therapeutic agents: a new perspective. In Alessio E (Ed.). Bioinorganic medicinal chemistry. John Wiley & Sons, Weinheim. 2011. pp. 197-222.
31. Gunatilleke SS, de Oliveira CA, McCammon JA, Barrios AM. Inhibition of cathepsin B by Au(I) complexes: a kinetic and computational study. J Biol Inorg Chem 2008;13:555-61.
32. Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE. AMBER 12. University of California, San Francisco, USA 2012.
33. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res 2000;28:235-42.
34. Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013;29:845-54.
35. Jain AN. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput Aided Mol Des 2007;21:281-306.
36. Gunatilleke SS. Inhibition of csteine proteases by gold(I) complexes: A kinetic and mechanistic investigation. PhD Dissertation, University of Southern California. Available from: http://digitallibrary.usc.edu/cdm/ref/collection/p15799coll127/id/529088. [Last accessed on 8 Aug 2018].
37. Hulkower KI, Butler CC, Linebaugh BE, Klaus JL, Keppler D, Giranda VL, Sloane BF. Fluorescent microplate assay for cancer cell-associated cathepsin B. Eur J Biochem 2000;267:4165-70.
38. Greenspan PD, Clark KL, Tommasi RA, Cowen SD, McQuire LW, Farley DL, van Duzer JH, Goldberg RL, Zhou H, Du Z, Fitt JJ, Coppa DE, Fang Z, Macchia W, Zhu L, Capparelli MP, Goldstein R, Wigg AM, Doughty JR, Bohacek RS, Knap AK. Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. J Med Chem 2001;44:4524-34.
39. Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Krüger FA, Light Y, Mak L, McGlinchey S, Nowotka M, Papadatos G, Santos R, Overington JP. The ChEMBL bioactivity database: an update. Nucleic Acids Res 2014;42:D1083-90.
40. Jones PG, Maddock AG, Mays MJ, Muir MM, Williams AF. Structure and bonding in gold(I) compounds. Part 2. Mössbauer spectrum of linear gold(I) complexes. J Chem Soc Dalton Trans 1977;15:1434-9.
41. Madeira JM, Gibson DL, Kean WF, Klegeris A. The biological activity of auranofin: implications for novel treatment of diseases. Inflammopharmacology 2012;20:297-306.
42. Madeira JM, Schindler SM, Klegeris A. A new look at auranofin, dextromethorphan and rosiglitazone for reduction of glia-mediated inflammation in neurodegenerative diseases. Neural Regen Res 2015;10:391-3.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Raevsky AV, Sharifi M, Pinchuk V, Klegeris A. In silico design of novel gold-phosphate containing compounds as selective inhibitors of cathepsin B in neuroinflammation. Neurosciences 2018;5:33. http://dx.doi.org/10.20517/2347-8659.2018.34
AMA Style
Raevsky AV, Sharifi M, Pinchuk V, Klegeris A. In silico design of novel gold-phosphate containing compounds as selective inhibitors of cathepsin B in neuroinflammation. Neuroimmunology and Neuroinflammation. 2018; 5: 33. http://dx.doi.org/10.20517/2347-8659.2018.34
Chicago/Turabian Style
Raevsky, Alexsey V., Mohsen Sharifi, Vasily Pinchuk, Andis Klegeris. 2018. "In silico design of novel gold-phosphate containing compounds as selective inhibitors of cathepsin B in neuroinflammation" Neuroimmunology and Neuroinflammation. 5: 33. http://dx.doi.org/10.20517/2347-8659.2018.34
ACS Style
Raevsky, AV.; Sharifi M.; Pinchuk V.; Klegeris A. In silico design of novel gold-phosphate containing compounds as selective inhibitors of cathepsin B in neuroinflammation. Neurosciences. 2018, 5, 33. http://dx.doi.org/10.20517/2347-8659.2018.34
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 0 clicks
Cite This Article 0 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.