



Pyridinic-N-Co single-atom catalysts unlock sustainable and efficient quinoline synthesis via hydrogen-transfer-coupled annulation
 0
0 Abstract
Developing sustainable and atom-economical hydrogen transfer routes for constructing pharmacologically valuable quinoline scaffolds from abundant alcohol feedstocks remains a significant challenge. Herein, a tailored pyridinic-nitrogen-coordinated cobalt (Co) single-atom catalyst (Co-N/C-U) is showcased, enabling the efficient synthesis of quinoline derivatives from inexpensive and readily available 2-nitrobenzyl alcohol and various secondary or primary alcohols via a cascade hydrogen transfer process followed by annulation. Characterization confirmed that Co-N/C-U contains atomically dispersed Co centers and exhibits exceptional catalytic activity, accessing quinolines with up to 98% yield across a broad substrate scope (47 examples) with a turnover number of up to 30,808, outperforming state-of-the-art catalytic systems. This strategy demonstrates scalability to gram-scale reactions and enables the synthesis of the Cavosonstat derivative, while the pronounced stability and reusability of the catalyst further underscores its promising potential for practical implementation. Mechanistic studies revealed that the pyridinic-N-Co moiety plays a dual role, where the isolated Co sites facilitate efficient hydrogen transfer, and the neighboring pyridinic-N atoms act as basic sites to promote the key Friedländer cyclization step. Density functional theory calculations revealed that the enhanced catalytic performance of Co-N/C-U originates from its optimized pyridinic-N-Co coordination environment. This work establishes a sustainable route to a wide range of quinolines, providing a foundation for the precise design of next-generation SACs for complex organic transformations.

Keywords
INTRODUCTION
Concerns about environmental pollution and resource scarcity have led to a strong demand for the development of efficient catalytic conversion strategies that enable the production of functional products from readily available feedstocks with an environmentally friendly and atom-economical approach[1]. Alcohols represent inexpensive precursors that are readily available through biomass conversion or fermentation processes. In recent years, the hydrogen transfer functionalization of abundant and sustainable alcohols as feedstocks has received considerable interest[2-5]. This strategy avoids the direct utilization of high-pressure molecular hydrogen, eliminates the demand for special equipment, such as autoclaves, and offers the advantages of safer operation, improved environmental compatibility, atom economy, and simpler handling [Figure 1A][6-8]. As a result, the development of effective and sustainable pathways to upgrade inexpensive, abundant alcohols into value-added fine chemicals represents an urgent priority[9-11].
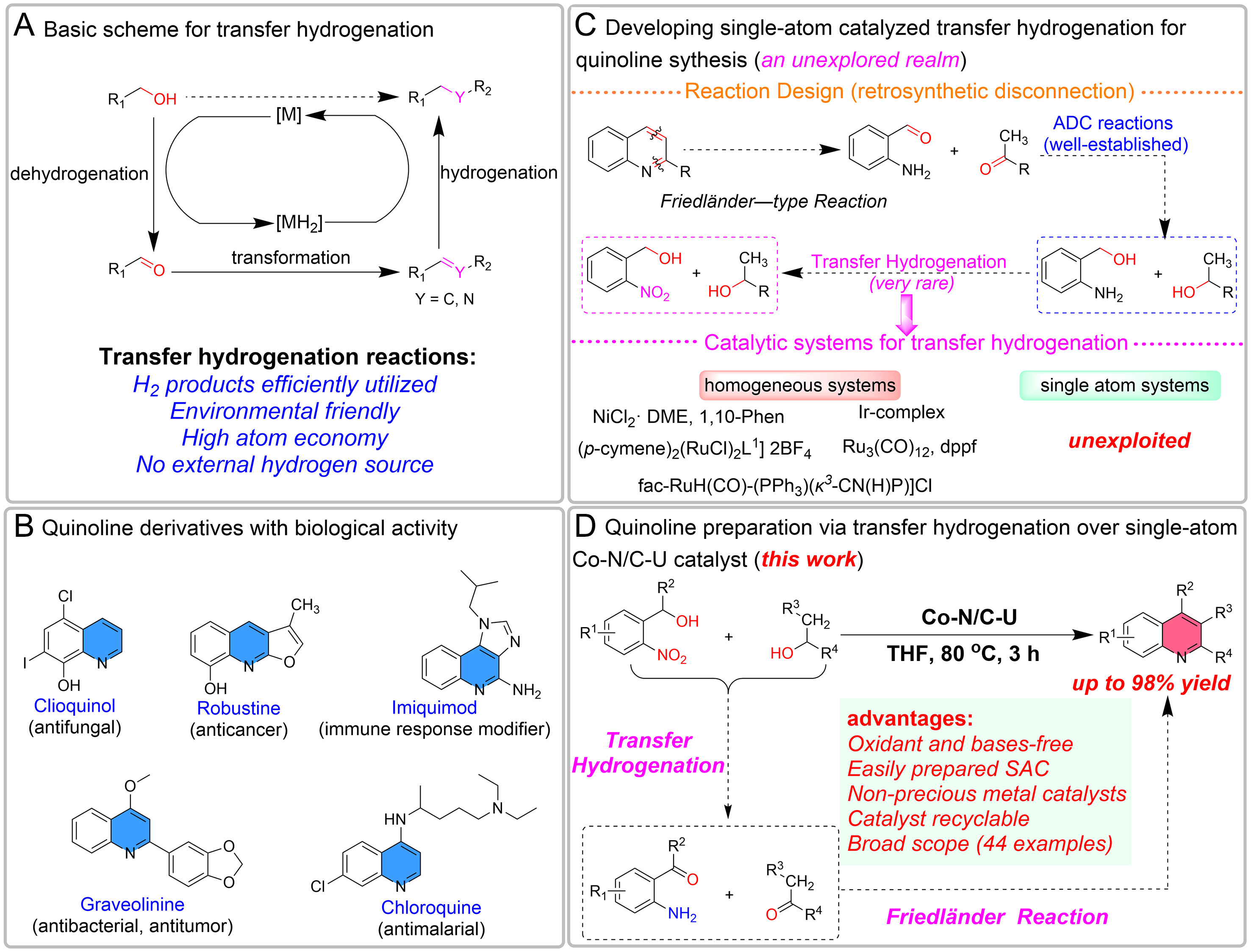
Figure 1. (A) Transfer hydrogenation reaction pathway; (B) quinoline products of biological activity; (C) transfer hydrogenation reaction designed for quinoline synthesis; and (D) synthesis of quinolines by transfer hydrogenation reaction over Co-N/C-U catalyst (this work).
Quinolines and their structural analogs constitute a prominent family of nitrogen-containing heterocycles, exhibiting diverse and potent biological profiles that include antitumor, antiviral, antimalarial, and anticancer properties [Figure 1B][12-17]. Furthermore, the latent diene and alkene subunits of the quinoline scaffold can be functionalized to assemble diverse bridged polycyclic compounds, establishing it as a valuable platform for constructing three-dimensional complex organic structures—an area of growing interest in medicinal chemistry[18,19]. Notwithstanding the development of a variety of synthetic methods for quinoline construction[20-24]—including the classical synthetic methods named after Skraup, Doebner, Doebner-von Miller, Combes, Camps, Pfitzinger, and Nimentowski—the Friedländer cyclization, developed over 135 years ago, remains one of the simplest and most direct approaches to obtaining quinolines[25]. However, the conventional protocol suffers from high temperatures and harsh conditions, as well as self-condensation of substrates such as 2-aminobenzaldehyde. To bypass substrate instability through modification or indirect Friedländer methods, the use of 2-aminobenzyl alcohol (US$127.63 per 500 g) instead of 2-aminobenzaldehyde derivatives as starting molecules provides a direct approach to synthesize quinoline via acceptorless dehydrogenative coupling (ADC) [Figure 1C][26-29]. This method has been extensively explored, and various catalytic systems have been developed, including homogeneous (Fe, Co, Ru, Mn complexes) and heterogeneous catalysts such as Pt, Ir, Co, and Mn nanoparticles[25,30-33]. However, these approaches often rely on precious metal catalysts and typically necessitate the addition of acid/base or redox reagents, while also suffering from challenges related to recyclability, complex precursor preparation, and low utilization of catalytic sites. In addition, these strategies mainly target the oxidation of alcohols, resulting in the waste of hydrogen from the dehydrogenation products with a reduced atom economy.
In recent years, another option has been to introduce a cheaper hydrogen-acceptor group, the nitro group, into the substrate 2-nitrobenzyl alcohol (US$69.34 per 500 g), which enables in situ transformation of the hydroxyl and nitro moieties into carbonyl and amino groups via hydrogen transfer, with the resulting species directly channeled into quinoline assembly [Figure 1C]. Environmentally and economically, 2-nitrobenzyl alcohol and secondary alcohols such as isopropanol represent low-cost and readily accessible raw materials, with water constituting the sole coproduct generated throughout the coupled cyclization and reduction steps. The in situ generation and consumption of reactive intermediates circumvent the need to isolate unstable species such as 2-aminobenzyl alcohol, aligning well with principles of sustainable synthesis[34]. Nevertheless, a key challenge in converting such 2-nitrophenyl alcohols and secondary alcohols into quinolines resides in the demand for a catalyst possessing sufficient activity to successively reduce the nitro group, thereby forming the essential amino functionality. At the same time, alcohol dehydrogenation must be deliberately and carefully controlled to stop at the aldehyde intermediate, since excessive dehydrogenation may lead to unwanted byproducts. Additionally, aldehydes are susceptible to aldol condensation, complicating the reaction. To our knowledge, only a handful of examples of quinoline synthesis via alcohol hydrogen transfer to reduce o-nitrobenzyl alcohol have been reported (listed in the Supplementary Figure 1)[35-41]. However, most of these catalysts are sensitive to air and/or moisture, and their synthesis process often complex, especially due to the complex ligands and precious metals, limiting industrial applications. Accordingly, designing innovative approaches to enable the direct and diversified sustainable production of quinolines using simple-to-prepare, reusable base-metal catalysts is highly desirable.
As an emerging paradigm in catalysis, single-atom catalysts (SACs) maximize atom utilization, enhance reaction efficiency, and offer superior stability and reusability, thereby merging the distinctive merits of homogeneous and heterogeneous systems[42,43]. In particular, single base metal (Co, Fe) atoms anchored within N-doped graphene matrices (M-N-C catalysts) have been intensively studied in thermal and electrochemical reactions[44-46]. Recently, such catalysts have been effectively used in organic coupling and hydrogenation reaction transformations[47,48], showing their great potential to substitute for costly noble-metal-based systems. Nevertheless, the M-N-C systems have not yet been applied to quinoline synthesis via hydrogen transfer coupling reactions. Moreover, while fine-tuning the carbon support can substantially boost activity via electron-metal-support interaction (EMSI) effects, the precise nature of their active sites remains ambiguous[49,50]. Limited research has been dedicated to elucidating the specific functions of M-N moieties possessing varied nitrogen configurations (e.g., pyridinic, pyrrolic, or graphitic N) in organic transformations[51]. The incomplete understanding of the precise structural identity of the active centers significantly impedes the widespread use of these earth-abundant catalytic systems. Herein, we present homogeneous pyridinic-N coordinated Co SACs (Co-N/C-U) prepared by pyrolysis of a ZIF-8 (zeolitic imidazolate framework-8) precursor using an in situ ammoniation engineering strategy with urea. The catalyst efficiently catalyzes the synthesis of quinolines from 2-nitrobenzyl alcohol and secondary alcohols via a cascade hydrogen transfer and annulation [Figure 1D]. Importantly, the Co-N/C-U SAC was air-stable and recyclable, with a broad substrate range (47 examples), and the scheme could be scaled up, which further demonstrated the practical applicability of this catalytic platform.
MATERIALS AND METHODS
Materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99.0%), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 99.0%), 2-methylimidazole(C4H6N2, 98%), urea (H2NCONH2, 99.5%), methanol (CH3OH, 99.5%), anhydrous ethanol (C2H5OH, 99.7%), tetrahydrofuran (THF, C4H8O, 99.0%), dimethyl sulfoxide (DMSO, C2H6SO, 99.0%), N, N-dimethylformamide (DMF, C3H7NO, 99.5%), Acetonitrile (CH3CN, 99.0%), 1,4-Dioxane (C4H8O2, 99.0%) were purchased from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). All reagents used for substrate scope expansion were also sourced from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Additionally, 2-Nitrobenzyl alcohol (C7H7NO3, 98.0%), Isopropanol (C3H8O, 99.7%), Cobalt(II) oxide(CoO, 99.0%), Ruthenium on carbon (Ru/C, Ru 5%), Palladium on carbon (Pd/C, Pd 10%), and Platinum on activated carbon (Pt/C, Pt 5%) were purchased from Sinopharm Group Co., Ltd. (Shanghai, China). All chemicals were used as received, without undergoing additional purifying processes.
Synthesis of Co-N/C-U catalyst
Typically, a methanol solution (120 mL) containing Co(NO3)2·6H2O (1.0 mmol, 0.29 g) and Zn(NO3)2·6H2O (11.0 mmol, 3.24 g) was prepared as solution A. Meanwhile, solution B was obtained by dissolving 2-methylimidazole (49.0 mmol, 4.02 g) and urea (245.0 mmol, 14.7 g) in methanol (80 mL). At room temperature, solution A was introduced dropwise to solution B over 30 min. The solid product was isolated by centrifugation, rinsed twice with ethanol, and dried overnight under vacuum at 60 °C to afford the ZnCo-bimetallic metal-organic framework (BMOF) precursor. Subsequently, this precursor powder was transferred into a tube furnace and pyrolyzed at 900 °C (heating rate: 5 °C·min-1) under a continuous argon stream. After holding at 900 °C for 3 h, the sample was cooled passively to ambient temperature, yielding the final Co-N/C-U material without further post-treatment.
Synthesis of Co-N/C catalyst
The synthesis of Co-N/C followed the same procedure as that for Co-N/C-U, except that urea was omitted from solution B.
Procedure for the quinoline synthesis
A mixture of 2-nitrobenzyl alcohol (0.5 mmol, 1.0 equivalent (equiv.)), isopropanol (1.1 mmol, 2.2 equiv.), and Co-N/C-U catalyst (5.0 mg) in THF (3.0 mL) was stirred under a nitrogen atmosphere in a 10 mL tube containing a magnetic stir bar. After heating at 80 °C for 3 h, the reaction mixture was cooled to room temperature. The catalyst was removed by centrifugation. The crude product was obtained by filtration through a Celite pad and subsequent concentration under reduced pressure, followed by final purification using flash column chromatography.
Characterization
X-ray diffraction (XRD) measurements were carried out using a SmartLab-SE diffractometer (Rigaku, Japan) with Cu Kα radiation (λ = 1.5418 Å). Scanning electron microscopy (SEM, ZEISS Sigma300, Germany) coupled with energy-dispersive X-ray spectroscopy (EDS, smart EDX) was employed to examine the surface morphology and elemental distribution of the samples. Microstructural and crystallographic analyses were conducted using transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) on a JEM-2100 instrument (JEOL Ltd., Tokyo, Japan). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were acquired on a JEM-2800 scanning transmission electron microscope (JEOL Ltd., Tokyo, Japan). Co K-edge extended X-ray absorption fine structure (EXAFS) spectra were collected in transmission mode using a Table XAFS-500A spectrometer (Specreation Instruments Co., Ltd., China). The elemental composition and chemical states of surface species were analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Fisher ESCALAB XI+, USA). Textural properties, including specific surface area and pore size distribution, were evaluated by nitrogen physisorption at 77 K based on the Brunauer-Emmett-Teller (BET) model (Micromeritics ASAP2020HD88, Micromeritics, USA). Raman spectra were obtained with a DXR confocal Raman spectrometer (Thermo Fisher Scientific, DXR3 SmartRaman, USA). Metal content in M-N-C was measured by an inductively coupled plasma optical emission spectrometer (ICP-OES, SpectroBlue, Germany).
Computational details
Spin-polarized density functional theory (DFT) calculations were performed in Gaussian 16 using the B3LYP (Becke 3-parameter Lee-Yang-Parr) hybrid GGA (generalized gradient approximation) exchange-correlation functional[52].The DFT-D3 method was applied to account for van der Waals interactions. All geometries were fully optimized, with energy calculations employing the def2-SVP basis set for all elements. The spin density analysis was carried out using the Multiwfn[53]and Visual Molecular Dynamics (VMD)[54] software tools, providing crucial insights into the molecular interactions and electronic properties of the studied systems.
RESULTS AND DISCUSSION
The Co-N/C-U SAC was synthesized according to an adjusted bimetallic organic framework self-adjustment strategy [Figure 2A]. A methanolic solution of cobalt nitrate and zinc nitrate was added to a second methanol solution containing 2-methylimidazole and urea (1:5), and the mixture was stirred for 3 h at room temperature. The resulting solid was isolated by centrifugation, washed, and dried to afford a pink precursor material.
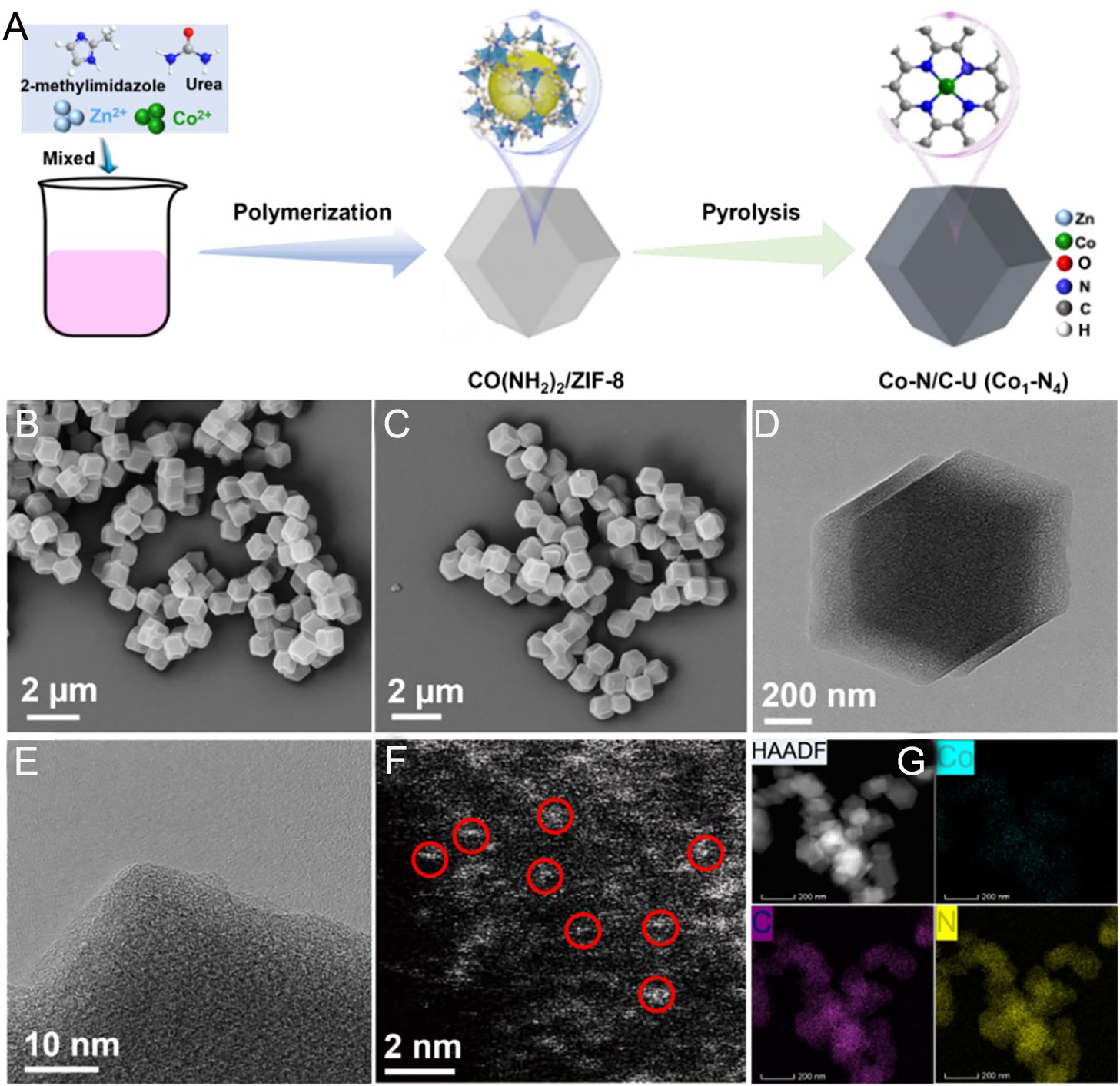
Figure 2. (A) Schematic illustration of the synthesis procedure of the Co-N/C-U catalyst. SEM images of (B) Co-N/C, and (C) Co-N/C-U. (D) TEM and (E) HRTEM images of Co-N/C-U. (F) AC HAADF-STEM image and (G) EDS elemental mappings of Co-N/C-U.
This precursor was finally calcined at 900 °C for 3 h in flowing argon to yield the Co-N/C-U catalyst. For comparison, the Co-N/C catalyst was synthesized following an identical protocol to that of Co-N/C-U, but omitting urea from the precursor mixture, [Supplementary Figure 2A]. As shown in the corresponding SEM images [Figure 2B and C], Co-N/C and Co-N/C-U both display a polyhedral structure, demonstrating that the urea treatment leaves the morphological framework intact. TEM and high-resolution TEM (HR-TEM) images of Co-N/C and Co-N/C-U [Figure 2D and E; Supplementary Figure 2B and C] show no evidence of cobalt clusters or nanoparticles, suggesting that Co species are predominantly atomically dispersed. This atomic-scale distribution was directly confirmed by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) [Figure 2F; Supplementary Figure 2D], in which individually bright dots correspond to isolated cobalt atoms. Furthermore, energy-dispersive X-ray spectroscopy (EDX) elemental maps of Co-N/C and Co-N/C-U confirm the homogeneous dispersion of Co, N, and C throughout the carbon matrix [Figure 2G; Supplementary Figure 3]. The cobalt loading in Co-N/C-U and Co-N/C was determined to be 2.33 and 2.31 wt%, respectively, by inductively coupled plasma optical emission spectrometry (ICP-OES).
The pyrolysis of 2-methylimidazole and urea generated abundant micropores and mesopores within the Co-N/C-U catalysts, as evidenced by BET measurements [Supplementary Figure 4]. N2 adsorption-desorption isotherms of Co-N/C-U displayed a characteristic type IV profile, indicative of a mesoporous framework, along with a type I regime at low relative pressure (P/P0 = 0-0.1) associated with microporosity. The total surface area was as high as 789.3 m2/g, significantly higher than that of Co-N/C.
Powder XRD patterns recorded for the as-synthesized Co-N/C and Co-N/C-U catalysts show a characteristic graphite (002) reflection at 26.38° and an absence of cobalt-related crystalline peaks, confirming high Co dispersion [Figure 3A]. A slight negative shift of this peak in Co-N/C-U relative to Co-N/C suggests that nitrogen incorporation expands the carbon lattice[55]. Raman spectroscopy was performed to analyze the carbon structures in Co-N/C and Co-N/C-U. As displayed in Figure 3B, two distinct bands are observed at around 1,336 and 1,593 cm-1, corresponding to the defective carbon (D band) and graphitic carbon (G band), respectively[56].The intensity ratio of D and G bands (ID/IG) increased from 0.91 (Co-N/C) to 1.14 (Co-N/C-U) for Co-N/C-U (1.14), indicating that the in situ urea pyrolysis introduces additional structural defects, which likely enhance the content of pyridinic- or pyrrolic-type nitrogen configurations. The chemical states of Co, N, and C in both Co-N/C and Co-N/C-U were analyzed by X-ray photoelectron spectroscopy (XPS). As shown in Figure 3C, the high-resolution Co 2p spectrum exhibits characteristic doublets with binding energies at approximately 781.0 and 796 eV, corresponding to the Co 2p3/2 and Co 2p1/2 peaks of Co2+, respectively, accompanied by satellite features between 785.1 and 803.7 eV. No discernible peak is observed at 778.3 eV, which excludes the presence of metallic Co particles or clusters[57,58]. Furthermore, the positive shift of the Co 2p3/2 peak from 780.8 to 781.0 eV confirms the coordination of Co(II) centers with nitrogen, forming Co-Nx moeities[58,59]. Notably, Co-N/C-U exhibits an ~0.90 eV higher Co 2p3/2 binding energy than Co-N/C (779.9 eV), consistent with a slightly more oxidized Co environment and stronger Co-N coordination. Deconvolution of the N 1s XPS spectra resolved five species at 397.9 eV (pyridinic nitrogen), 398.6 eV (Co-Nx), 399.2 eV (pyrrolic nitrogen), 400.7 eV (graphitic nitrogen), and
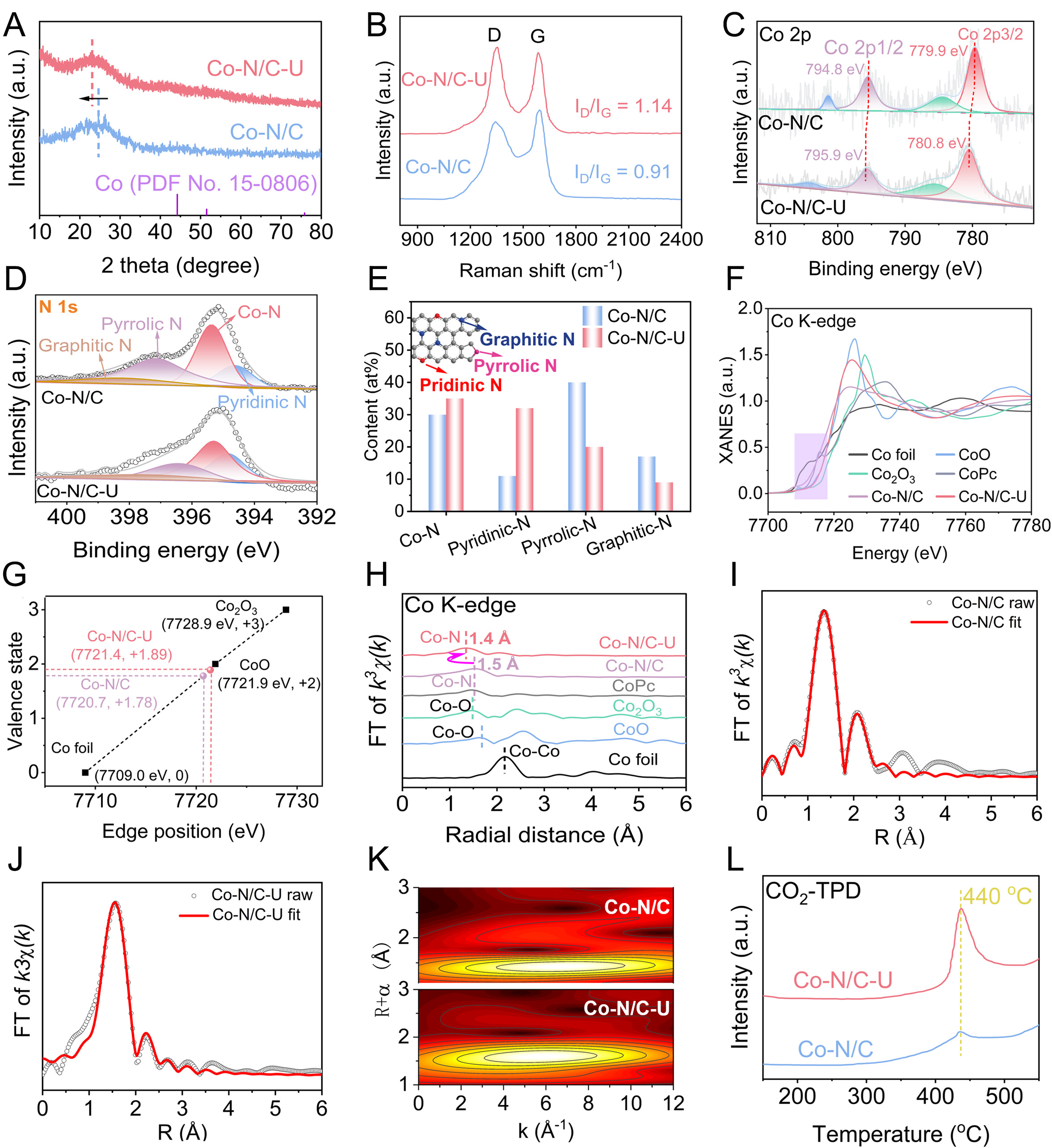
Figure 3. (A) XRD patterns, (B) Raman spectra, (C) XPS envelopes of (C) Co 2p and (D) N 1s, and (E) proportion of different N species determined by XPS analysis of Co-N/C and Co-N/C-U. (F) Co K-edge XANES spectra, (G) Relationship between the valence states of Co and energy position of XANES spectra, and (H) FT k3-weighted Co K-edge EXAFS spectra of Co-N/C, Co-N/C-U, and reference samples. (I and J) Co K-edge EXAFS curve fitting results R space, (K) Wavelet transform (WT) of Co-N/C and Co-N/C-U, and (L) CO2-TPD profiles of Co-N/C and Co-N/C-U.
X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) provided further insight to elucidate the electronic structure and the microenvironment around isolated Co sites in the Co-N/C-U catalyst. The normalized Co K-edge XANES spectra of Co-N/C and Co-N/C-U revealed that the absorption edge positions of both Co-N/C and Co-N/C-U are close to that of CoO but distinct from those of Co foil and Co2O3, indicating a similar oxidation state of cobalt in these samples as in CoO [Figure 3F][65]. Fitted XANES results estimated average Co oxidation states of +1.78 for Co-N/C and +1.89 for Co-N/C-U [Figure 3G], which is consistent with the corresponding Co 2p XPS spectral analysis. The Fourier transform (FT) k3-weighted EXAFS spectra of the Co k-edge in Figure 3H featured a predominant peak at ~1.4-1.5 Å for both Co-N/C and Co-N/C-U, corresponding to Co-N coordination, while no peak attributable to Co-Co bond is observed at ~2.18 Å[66]. The results indicate that the Co atoms are exclusively coordinated with the N atoms in the nitrogen-carbon support, with no formation of Co nanoparticles or clusters, consistent with the observations from aberration-corrected (AC)-HAADF-STEM imaging. The least-square EXAFS fitting results for Co in R-space and K-space and the extracted structural parameters clearly showed that the coordination number of Co in both Co-N/C and Co-N/C-U is close to four [Figure 3I and J, Supplementary Figures 6 and 7, Supplementary Table 1]. The local Co environment in Co-N/C and Co-N/C-U was further analyzed by EXAFS wavelet transform (WT) [Figure 3K]. The peak intensity centered at ~5 Å corresponds closely to that of Co-Pc, a result that is consistent with Co-N bonding[67], and no intensity maximum corresponding to Co-Co was observed by comparing to the WT plots of the other reference samples (Co foil, CoO) [Supplementary Figure 8]. Compared with Co-N/C, the CO2-temperature-programmed desorption (CO2-TPD) profile further demonstrates that the urea-treated catalyst (Co-N/C-U) possesses a significantly higher density of medium- to strong-strength basic sites [Figure 3L]. This result corroborates that the high-density pyridinic N sites are the predominant contributor to the catalyst's basicity. Based on these characterization results, urea plays a dual role in both the synthesis and catalytic performance of Co-N/C-U. During the pyrolysis process, urea acts as an additional nitrogen source, effectively increasing the pyridinic nitrogen content within the carbon matrix. These pyridinic nitrogen species preferentially coordinate with cobalt atoms to form the catalytically active pyridinic-N-Co motifs. The resulting pyridinic-N-Co moieties not only stabilize the isolated cobalt centers and modulate their electronic structure but also provide abundant basic sites. These combined features collectively contribute to the efficient promotion of subsequent catalytic reactions.
Hydrogen transfer reactions of alcohols represent an attractive route for the sustainable and economical synthesis of quinoline heterocycles, as alcohols are readily available precursors and water is the sole byproduct. Despite their promise, most reported systems rely on homogeneous catalysts, and only a limited number of heterogeneous examples exist. Applying heterogeneous Co SACs to this reaction not only provides novel and more efficient catalysts for this transformation but also provides mechanistic insight into hydrogen transfer and ringclosing pathways. The catalytic activity of Co-N/C-U was first evaluated in the model reaction for synthesizing 2-methylquinoline (4a) from 2-nitrobenzyl alcohol (1a) and isopropanol (2a) [Figure 4A]. In this transformation, the Co-N/C-U catalyst showed outstanding catalytic performance under mild conditions (80 °C) without oxidants or bases, achieving quantitative conversion of the target product 2-methylquinoline within 3 h with > 98% yield. By contrast, Co-N/C (without urea) yielded only 38% of the target product. To assess the contribution of metal single atoms to the activity, a metal-free N-doped counterpart carbon framework (N/C) was synthesized following the same procedure using 2-methylimidazole and urea. Although metal-free N-doped carbon materials are reported to catalyze certain dehydrogenation reactions[68-70],the N/C material exhibited no detectable catalytic activity under the present model reaction conditions. Interestingly, the coordination behavior of the Co-N4 containing phthalocyanine macromolecule (Co-PC) with a structure analogous to that of the Co-N/C catalyst also showed negligible activity. Moreover, treating Co-N/C-U with potassium thiocyanate (KSCN) significantly suppressed its catalytic performance (20% yield), as thiocyanate (SCN-) strongly binds to cobalt centers and blocks the active sites[71]. These findings suggest that both the microenvironment of the Co-N4 moiety and the basicity of the N-doped carbon materials are critical for both efficient alcohol dehydrogenation and hydrogen borrowing coupled cyclization. Unsurprisingly, conventional cobalt-based compounds (e.g., CoO and Co(NO3)2·6H2O) as well as supported noble-metal catalysts (e.g., Pt/C, Pd/C, and Ru/C) showed little to no activity in the reaction of 2-nitrobenzyl alcohol with isopropanol to obtain target compound 4a.
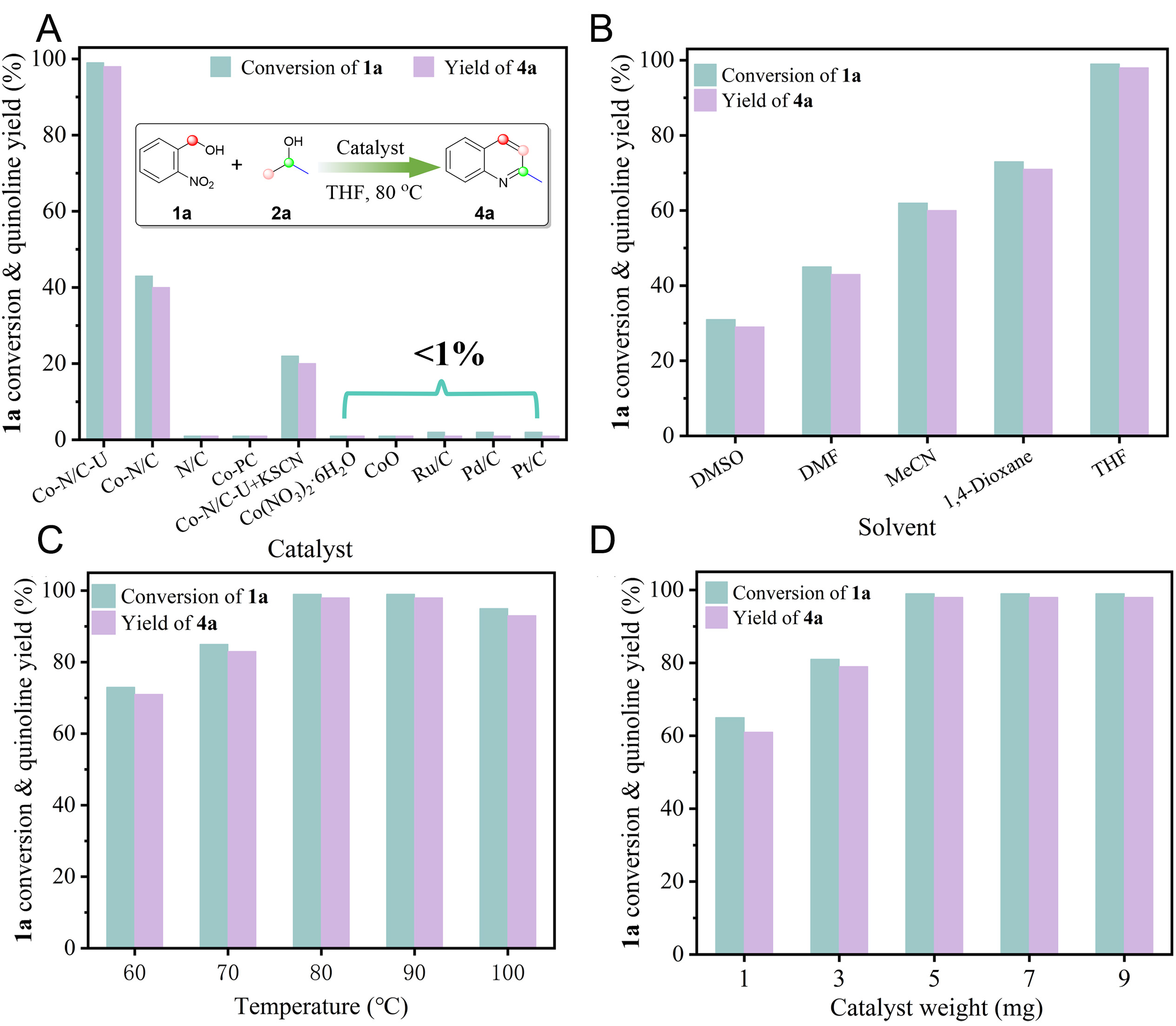
Figure 4. (A) Catalyst evaluation test, (B) the solvent effect, (C) the influence of temperature, and (D) the Co-N/C-U catalyst amount effect. Reaction conditions: 2-nitrobenzyl alcohol 1a (0.5 mmol), isopropanol 2a (1.1 mmol), solvent or THF (3 mL), catalyst (0.4 mol% metal or Co), N2, 80 °C, and 3 h. The yield and conversion are determined by GC using hexadecane as the internal standard.
Subsequently, we investigated the effect of the reaction solvent [Figure 4B]. The properties of the solvent, such as polarity, significantly affect this transformation. Lower yields (25%-42%) were observed in strongly polar solvents such as DMSO and DMF. Yields increased sharply as solvent polarity decreased, reaching 58% and 71% for 4a in acetonitrile and 1,4-dioxane, respectively. THF was identified as the optimal solvent, yielding quantitative amounts of target product 4a. The effect of reaction temperature was systematically evaluated. The reaction was found to be optimal at 80 °C, affording a 98% yield. Lowering the temperature from 80 to 60 °C decreased the yield from 98% to 72%, whereas raising it to 100 °C provided no further improvement and instead caused a slight decline [Figure 4C]. Optimization of the catalyst (Co-N/C-U) loading revealed that 5 mg was the optimal dosage [Figure 4D].
With optimal conditions in hand, we probed the universality of this synthetic approach [Figure 5]. Mono-substituted nitrobenzyl alcohol with various electron-donating, electron-drawing, and neutral groups (-Me, -OMe, -OCH2CH3, -NH2, -N(CH3)2, -OH, -CF3, -F, -Cl, -Br, -COOCH3) all reacted smoothly to give the corresponding quinoline compounds 4b-4r with up to 97% yield. The electronic nature of the substituents exerted a subtle yet noticeable influence, with electron-donating groups generally leading to higher yields than electron-withdrawing groups (4c-4e & 4k-4m). Substituent position also affected reactivity.
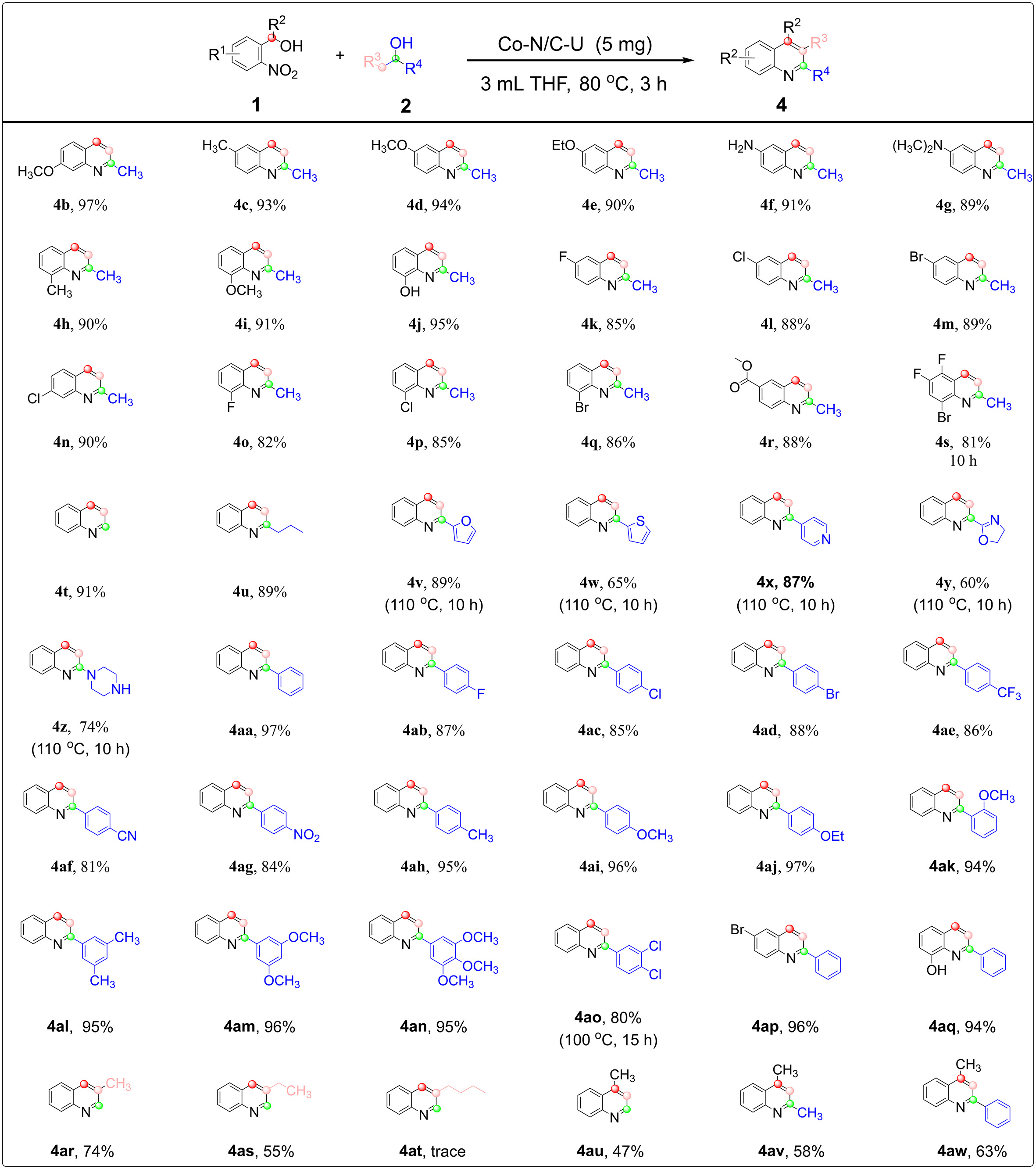
Figure 5. Co-N/C-U catalyzed tandem hydrogen transfer-cyclization for the synthesis of quinolines. Standard conditions: 2-nitrobenzyl alcohol (1, 0.5 mmol), alcohol (2, 1.1 mmol), Co-N/C-U (5 mg), THF (3.0 mL), at 80 °C for 3 h.
This is illustrated by methoxy-substituted nitrobenzyl alcohols, where the reactivity decreases in the order 4-methoxy > 5-methoxy > 3-methoxy isomers, a trend notably governed by increasing steric hindrance near the reaction center (4b & 4d & 4i). In addition, multi-halogen substituted nitrobenzyl alcohols ((3-bromo-5,6-difluoro-2-nitrophenyl)methanol) also gave the desired quinoline 4s smoothly in 82% yield by suitably extending the reaction time to 10 h, attributed to the marked electron-withdrawing character of the halogen substituents. Primary alcohols, with ethanol being a prominent example, exhibit higher inertness compared to isopropanol. Thus, ethanol dehydrogenation remains difficult even with precious-metal catalysts. Remarkably, the Co-N/C-U catalyst enabled efficient dehydrogenative coupling of nitrobenzyl alcohol with short-chain aliphatic alcohols, including ethanol and 2-pentanol, affording unsubstituted quinoline (4t) and 2-propylquinolines (4u) in yields of 91% and 89%, respectively. More importantly, heterocyclic alcohols were also viable substrates, releasing quinoline compounds with furan, thiophene, pyridine, oxazole, and morpholine moieties in yields ranging from 60% to 89% (4v-4z). Owing to the higher dehydrogenation energy barrier of heterocyclic alcohols, higher reaction temperature (110 °C) and long reaction time (10 h) are required to achieve the dehydrogenation coupling with nitrobenzyl alcohols. The substrate scope was further extended to the dehydrocoupling of 1-phenylethanol with 2-nitrobenzyl alcohol. Various 1-phenylethanol derivatives underwent successful dehydrocoupling with nitrobenzyl alcohol, affording the corresponding quinolines in 81%-97% yields (4aa-4ao). The electronic properties of the substituent groups of the 1-phenylethanol derivatives had a slight effect on the yields, with the observation that 1-phenylethanol with the electron-donating group was more active than that with an electron-withdrawing group (4ab-4ag & 4ah-4an & 4ao). Steric hindrance of the substituent group had little effect on the reaction efficiency (4aa & 4an). The procedure was successfully extended to the dehydrogenative coupling of substituted nitrobenzyl alcohols with 1-phenylmethanol to access quinolines (4ap, 4aq). Furthermore, this method proved to be applicable to the synthesis of both 3- and 4-substituted quinolines. When propanol and butanol were employed as coupling partners, the corresponding 3-substituted quinoline products, 3-methylquinoline (4ar) and 3-ethylquinoline (4as), were obtained in moderate to good yields of 74% and 55%, respectively, indicating that longer-chain alcohols exhibit lower reactivity in this transformation. Notably, when 1-(2-nitrophenyl)ethanol was subjected to the standard reaction conditions, this strategy enabled the construction of a series of 4-substituted quinoline derivatives, affording the target products in moderate to good yields (4au, 47%; 4av, 58%; 4aw, 63%).
To further confirm the practical utility of this protocol, the durability and recyclability of Co-N/C-U were examined in the model reaction. The catalyst retained its activity over six consecutive cycles without discernible loss, as shown in Figure 6A. Furthermore, hot filtration experiments showed that the reaction stopped after separating the Co-N/C-U catalyst, and the Co content in the solution phase was undetectable (detection limit < 10-4), indicating the absence of cobalt leaching [Supplementary Figure 9]. The spent Co-N/C-U catalyst was subsequently characterized. XRD measurements confirmed the structural integrity of the carbon matrix, with no characteristic diffraction peaks corresponding to Co clusters or nanoparticles [Supplementary Figure 10]. TEM images further confirmed that the spent catalyst largely retained the original dodecahedral morphology. The AC-HAADF-STEM images indicated that Co species were maintained predominantly as isolated single atoms, with no apparent aggregation into nanoparticles or clusters [Supplementary Figure 11]. The Co 2p XPS spectra of the spent Co-N/C-U catalyst were nearly identical to those of the fresh sample. The two fitted peaks at 781.0 and 796 eV confirmed that the Co-N4 motif remained intact after the reaction [Supplementary Figure 12]. Combined evidence from recycling tests and post-reaction characterization demonstrates the exceptional stability of the spent Co-N/C-U catalysts.
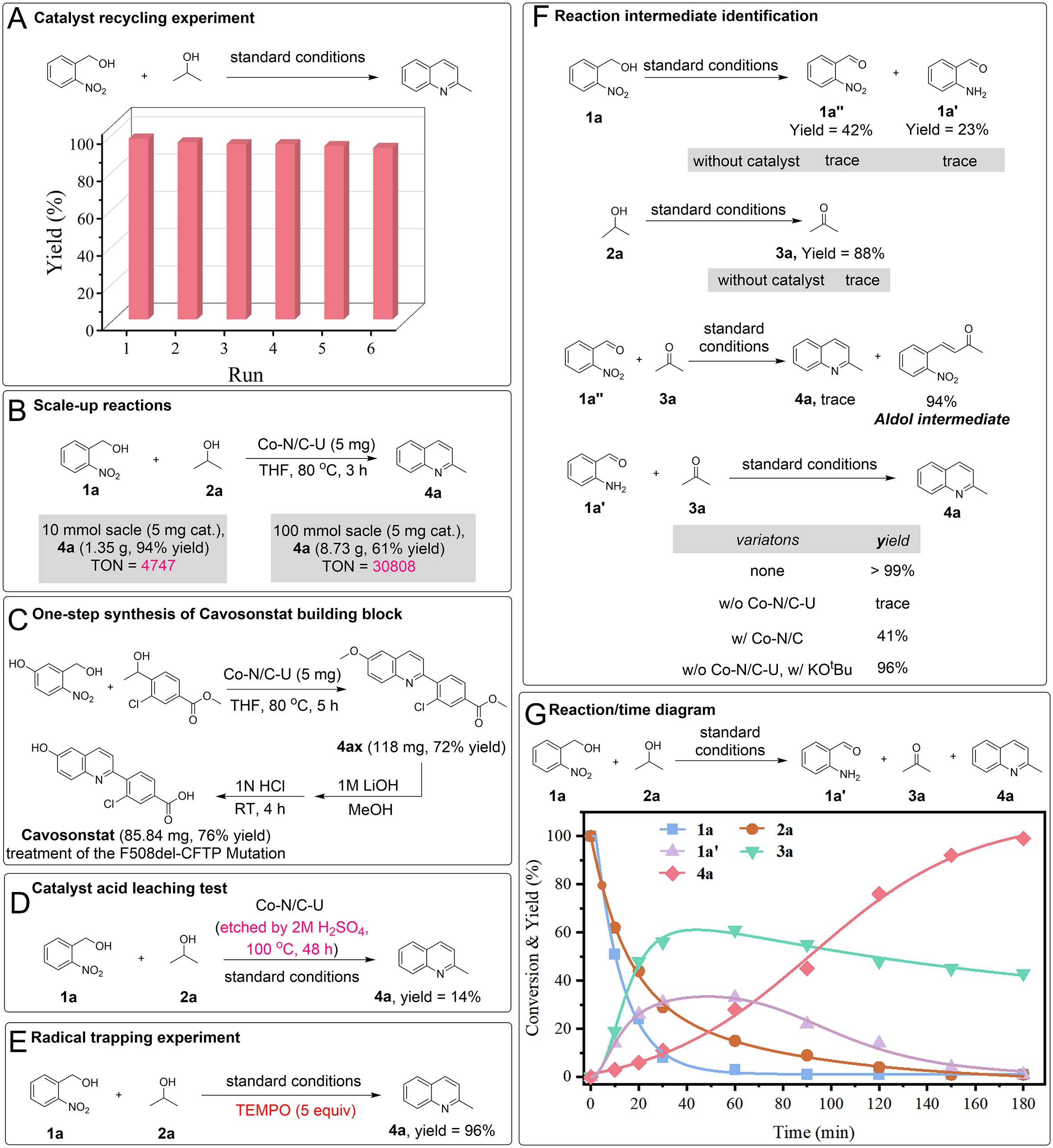
Figure 6. (A) Catalyst recycling experiment, (B) scale-up reactions, (C) one-step synthesis of Cavosonstat building block, (D) acid-etching experiment, (E) radical inhibition experiment, (F) identification of intermediates, and (G) reaction/time diagram.
To demonstrate the practical utility of this system, the Co single-atom-catalyzed hydrogen transfer strategy was successfully applied to gram-scale synthesis and the construction of specific target molecules. For the coupling reaction of 1a and 2a using 5 mg of the Co-N/C-U catalyst under N2, a 10 mmol scale reaction afforded 4a in 94% yield with a turnover number (TON) of 4,747. Under identical catalyst loading, the
A series of mechanistic experiments was performed to elucidate the reaction pathway. The identity of the active sites was probed by acid leaching, where treating Co-N/C with 1 M H2SO4 at 100 °C for 48 h removed most isolated cobalt species [Figure 6D], and the resulting material produced only 14% of 4a. The result implicates the isolated Co centers as critical active sites, which aligns with the findings from the KSCN test [Figure 4A]. The addition of 2,2,6,6-tetramethylpiperidine N-oxyl (TEMPO) showed no inhibitory effect on the reaction between 1a and 2a under the standard conditions, suggesting that the reaction process probably does not involve radical participation [Figure 6E].
To gain further mechanistic insight into the reaction, the individual dehydrogenation reactions of 1a and 2a were examined separately under standard conditions [Figure 6F]. Substrate 1a produced two carbonyl compounds: 2-nitrobenzaldehyde (1a'', 42% yield) and 2-aminobenzaldehyde (1a', 23% yield), the latter arising from intramolecular hydrogen transfer. Concurrently, 2a was converted to acetone (3a) in up to 88% yield. Control experiments confirmed that both dehydrogenation pathways were strictly dependent on the Co-N/C-U catalyst, as negligible product formation occurred in its absence. Subsequent condensation tests identified the key reactive intermediates [Figure 6F]. When 1a'' was used in place of 1a for condensation with 3a, quinoline formation was negligible. In contrast, 1a' reacted smoothly with 3a, delivering 4a in 99% yield, thus establishing 1a' and 3a as the essential intermediates in the transformation. In the absence of Co-N/C-U, 1a' and 3a did not form 4a, and the yield dropped to 41% when Co-N/C-U was replaced by Co-N/C. Notably, the reaction proceeded efficiently (96% yield) when only KOtBu was added as a base, demonstrating that the pyridinic-N sites in the catalyst provide the necessary basicity for the condensation, whereas the Co single-atom centers are not directly involved in this specific transformation step.
The time-dependent concentration profiles of 1a, 2a, and key intermediates were further analyzed
To elucidate the mechanism underlying the enhanced catalytic activity imparted by the developed catalyst (Co-N/C-U), we performed systematic DFT calculations to analyze the electronic structures, substrate adsorption behaviors, and reaction energetics for the Co-N/C-U and Co-N/C catalysts. First, structural optimization yielded two representative catalyst models featuring pyridinic-nitrogen (Co-N/C-U) and pyrrolic-nitrogen (Co-N/C), respectively [Figure 7A]. Adsorption energy calculations reveal a distinct difference in substrate affinity between the two catalysts (Co-N/C-U and Co-N/C). The adsorption of 1a on Co-N/C-U (-9.89 kcal/mol) is significantly weaker than that on Co-N/C (-16.49 kcal/mol), whereas both catalysts exhibit comparable adsorption strength toward 2a [Figure 7A]. This differential affinity suggests that, after activation, 1a can desorb more readily from Co-N/C-U, thereby potentially promoting the overall reaction turnover. Given the well-established mechanism for the isopropanol-mediated hydrogen-borrowing reduction of nitro groups and subsequent Friedländer cyclization to form quinolines, we focus our analysis on the key intramolecular O-H hydrogen transfer step for nitro reduction within 1a [Figure 7B]. The corresponding reaction energy profiles indicate that on the Co-N/C-U catalyst, the process proceeds via a transition state (TS1) with a barrier of 24.63 kcal/mol, leading to a hydrogen transfer product (P1) and exhibiting a more favorable thermodynamic profile. In contrast, on the Co-N/C catalyst, despite the formation of an exceptionally stable initial adsorption complex (Co-N/C-1a, -16.49 kcal/mol), the subsequent hydrogen-transfer step transition state (TS2) still presents a high barrier (TS2 = 36.61 kcal/mol). This demonstrates that overly strong adsorption on Co-N/C does not translate into kinetic advantage and may instead result in active-site saturation. Spin density analysis further elucidates the electronic effect of the differing coordination environments of the catalyst [Figure 7C]. In the pristine catalysts, the spin density in Co-N/C-U is substantially delocalized between the Co center and its surrounding N/C matrix, whereas it is highly localized on the Co atom in Co-N/C. This distinction is crucial for substrate adsorption and activation [Supplementary Table 2]. For instance, in Co-N/C-U-1a and Co-N/C-U-2a, a portion of the spin density transfers to relevant atoms of the adsorbates, indicating charge redistribution that likely facilitates hydrogen atom transfer. Overall, these results demonstrate that the Co-N/C-U catalyst achieves a more efficient catalytic cycle by modulating the metal coordination environment to optimize the active-site electronic structure, thereby promoting spin density delocalization, moderating substrate adsorption strength, and stabilizing key transition states. This modification strategy successfully circumvents the kinetic limitations arising from excessively strong adsorption on Co-N/C, thus balancing the activation and desorption events and ultimately enhancing the overall catalytic efficiency.

Figure 7. (A) Optimized adsorption structures and calculated adsorption energies (Eads) for isopropanol on Co-N/C-U and Co-N/C surfaces. (B) Reaction energy profiles for the intramolecular hydroxy hydrogen transfer in 2-nitrobenzyl alcohol over Co-N/C-U and Co-N/C. (C) Spin-density isosurfaces (isovalue = 0.05) for Co-N/C-U and Co-N/C. O, Co, C, N, and H atoms of the optimized intermediate structures are displayed in red, purple, gray, blue, and white, respectively.
CONCLUSIONS
This study aimed to develop a sustainable and efficient catalytic system for quinoline synthesis via hydrogen transfer-coupled annulation. A tailored cobalt SAC with pyridinic-N coordination (Co-N/C-U) was successfully fabricated, and it exhibited exceptional activity, broad substrate scope, and excellent recyclability in this transformation. Mechanistic investigations revealed the dual role of the pyridinic-N-Co moiety, wherein the isolated Co centers facilitate hydrogen transfer while adjacent pyridinic-N atoms promote Friedländer cyclization. This work provides a fundamental understanding of structure-activity relationships in M-N-C SACs, paving the way for their rational design and application in complex organic transformations. Overall, the insights gained from this work lay a strong foundation for advancing sustainable catalytic strategies for quinoline synthesis using earth-abundant single-atom systems.
DECLARATIONS
Authors’ contributions
Conceived and designed the study, manuscript revision and supervising: Li, H.; Yang, S.
Prepared the samples and collected the data, performed data analysis and wrote the main draft of the paper: Xu, F.; Zhang, L. L.
All authors participated in the writing of the manuscript.
Availability of data and materials
The data supporting the findings of this study are available within this Article and its Supplementary Materials. According to reasonable requirements, all the data examined in this research can be obtained from the correspondents.
AI and AI-assisted tools Statement
Not applicable.
Financial support and sponsorship
This work is financially supported by the Guizhou Provincial S&T Project (QianKeHePingTaiCXPTXM(2025)023, ZK[2022]011) and the Doctoral Research Start-up Fund of Guiyang University (Grant No. GYU-KY-[2026]-01).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Garg, N.; Sarkar, A.; Sundararaju, B. Recent developments on methanol as liquid organic hydrogen carrier in transfer hydrogenation reactions. Coord. Chem. Rev. 2021, 433, 213728.
2. Jafarzadeh, M.; Sobhani, S. H.; Gajewski, K.; Kianmehr, E. Recent advances in C/N-alkylation with alcohols through hydride transfer strategies. Org. Biomol. Chem. 2022, 20, 7713-45.
3. Yan, H.; Liao, Q.; Chen, Y.; et al. Photocatalytic metal hydride hydrogen atom transfer mediated allene functionalization by cobalt and titanium dual catalysis. Angew. Chem. Int. Ed. 2023, 135, e202302483.
4. Gao, Y.; Hong, G.; Yang, B. M.; Zhao, Y. Enantioconvergent transformations of secondary alcohols through borrowing hydrogen catalysis. Chem. Soc. Rev. 2023, 52, 5541-62.
5. Reed-Berendt, B. G.; Latham, D. E.; Dambatta, M. B.; Morrill, L. C. Borrowing hydrogen for organic synthesis. ACS. Cent. Sci. 2021, 7, 570-85.
6. Cao, H.; Tang, X.; Tang, H.; Yuan, Y.; Wu, J. Photoinduced intermolecular hydrogen atom transfer reactions in organic synthesis. Chem. Catal. 2021, 1, 523-98.
7. Yang, C.; Arora, S.; Maldonado, S.; Pratt, D. A.; Stephenson, C. R. J. The design of PINO-like hydrogen-atom-transfer catalysts. Nat. Rev. Chem. 2023, 7, 653-66.
8. Ang, H. T.; Miao, Y.; Ravelli, D.; Wu, J. Pyridine N-oxides as hydrogen atom transfer reagents for site-selective photoinduced Csp3-H functionalization. Nat. Synth. 2024, 3, 568-75.
9. Nallagangula, M.; Subaramanian, M.; Kumar, R.; Balaraman, E. Transition metal-catalysis in interrupted borrowing hydrogen strategy. Chem. Commun. 2023, 59, 7847-62.
10. Podyacheva, E.; Afanasyev, O. I.; Vasilyev, D. V.; Chusov, D. Borrowing hydrogen amination reactions: a complex analysis of trends and correlations of the various reaction parameters. ACS. Catal. 2022, 12, 7142-98.
11. Li, J.; Mao, A.; Yao, W.; Zhu, H.; Wang, D. Iridium supported on porous polypyridine-oxadiazole as high-activity and recyclable catalyst for the borrowing hydrogen reaction. Green. Chem. 2022, 24, 2602-12.
12. Muthukrishnan, I.; Sridharan, V.; Menéndez, J. C. Progress in the chemistry of tetrahydroquinolines. Chem. Rev. 2019, 119, 5057-191.
13. Prabagar, B.; Yang, Y.; Shi, Z. Site-selective C-H functionalization to access the arene backbone of indoles and quinolines. Chem. Soc. Rev. 2021, 50, 11249-69.
14. Jeanmart, S.; Edmunds, A. J. F.; Lamberth, C.; Pouliot, M.; Morris, J. A. Synthetic approaches to the 2015-2018 new agrochemicals. Bioorg. Med. Chem. 2021, 39, 116162.
15. Yang, G. Z.; Zhu, J. K.; Yin, X. D.; et al. Design, synthesis, and antifungal evaluation of novel quinoline derivatives inspired from natural quinine alkaloids. J. Agric. Food. Chem. 2019, 67, 11340-53.
16. Afzal, O.; Kumar, S.; Haider, M. R.; et al. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871-910.
17. Kaur, K.; Jain, M.; Reddy, R. P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245-64.
18. Ma, J.; Chen, S.; Bellotti, P.; et al. Photochemical intermolecular dearomative cycloaddition of bicyclic azaarenes with alkenes. Science 2021, 371, 1338-45.
19. Yang, J.; Yang, L.; Zhao, Y.; et al. Asymmetric dearomative [2 + 2] photocycloaddition of quinoline and indole derivatives with Bicyclo[1.1.0]butanes. J. Am. Chem. Soc. 2025, 147, 35755-66.
20. Jia, H.; Tan, Z.; Zhang, M. Reductive functionalization of pyridine-fused N-heteroarenes. Acc. Chem. Res. 2024, 57, 795-813.
21. Kaur, R.; Kumar, K. Synthetic and medicinal perspective of quinolines as antiviral agents. Eur. J. Med. Chem. 2021, 215, 113220.
22. Patel, A.; Patel, S.; Mehta, M.; et al. A review on synthetic investigation for quinoline- recent green approaches. Green. Chem. Lett. Rev. 2022, 15, 337-72.
23. Batista, V. F.; Pinto, D. C. G. A.; Silva, A. M. S. Synthesis of quinolines: a green perspective. ACS. Sustain. Chem. Eng. 2016, 4, 4064-78.
24. Mandal, A.; Khan, A. T. Recent advancement in the synthesis of quinoline derivatives via multicomponent reactions. Org. Biomol. Chem. 2024, 22, 2339-58.
25. Chelucci, G.; Porcheddu, A. Synthesis of Quinolines via a metal-catalyzed dehydrogenative N-heterocyclization. Chem. Rec. 2017, 17, 200-16.
26. Lu, Y.; Zhu, M.; Chen, S.; et al. Single-atom Fe-catalyzed acceptorless dehydrogenative coupling to quinolines. J. Am. Chem. Soc. 2024, 146, 23338-47.
27. Mastalir, M.; Glatz, M.; Pittenauer, E.; Allmaier, G.; Kirchner, K. Sustainable synthesis of quinolines and pyrimidines catalyzed by manganese PNP pincer complexes. J. Am. Chem. Soc. 2016, 138, 15543-6.
28. Yu, K.; Chen, Q.; Liu, W. Iron-catalysed quinoline synthesis via acceptorless dehydrogenative coupling. Org. Chem. Front. 2022, 9, 6573-8.
29. Tian, D.; He, Y. P.; Yang, L. S.; Li, Z. C.; Wu, H. Switchable skeletal editing of quinolines enabled by cyclizative sequential rearrangements. Nat. Chem. 2025, 17, 952-60.
30. Trincado, M.; Bösken, J.; Grützmacher, H. Homogeneously catalyzed acceptorless dehydrogenation of alcohols: a progress report. Coord. Chem. Rev. 2021, 443, 213967.
31. Crabtree, R. H. Homogeneous transition metal catalysis of acceptorless dehydrogenative alcohol oxidation: Applications in hydrogen storage and to heterocycle synthesis. Chem. Rev. 2017, 117, 9228-46.
32. Nguyen, D. H.; Trivelli, X.; Capet, F.; Paul, J.; Dumeignil, F.; Gauvin, R. M. M Manganese pincer complexes for the base-free, acceptorless dehydrogenative coupling of alcohols to esters: development, scope, and understanding. ACS. Catal. 2017, 7, 2022-32.
33. Pal, D.; Mondal, A.; Sarmah, R.; Srimani, D. Designing cobalt(II) complexes for tandem dehydrogenative synthesis of quinoline and quinazoline derivatives. Org. Lett. 2024, 26, 514-8.
34. Paul, B.; Panja, D.; Kundu, S. Synthesis of N-heterocycles through alcohol dehydrogenative coupling. Nat. Protoc. 2024, 19, 3640-76.
35. Chun, S.; Reddy Putta, R.; Hong, J.; Choi, S. H.; Oh, D. C.; Hong, S. Iron‐catalyzed transfer hydrogenation: Divergent synthesis of quinolines and quinolones from ortho‐nitrobenzyl alcohols. Adv. Synth. Catal. 2023, 365, 3367-74.
36. Xie, F.; Zhang, M.; Chen, M.; Lv, W.; Jiang, H. Convenient synthesis of quinolines from α‐2‐nitroaryl alcohols and alcohols via a ruthenium‐catalyzed hydrogen transfer strategy. ChemCatChem 2014, 7, 349-53.
37. Sk, M.; Bera, A.; Banerjee, D. Nickel‐catalyzed sequential dehydrogenation and cyclization of 2‐amino (nitro)‐benzyl alcohols with alkyl alcohols: synthesis of C‐3‐substituted auinolines. ChemCatChem 2023, 15, e202300412.
38. Zhao, L.; Chen, Y.; Zhang, C.; et al. Ru-CNP complex-catalyzed hydrogen transfer/annulation reaction of 2-nitrobenzylalcohol via an outer-sphere mechanism. J. Org. Chem. 2025, 90, 4959-72.
39. Maji, M.; Chakrabarti, K.; Panja, D.; Kundu, S. Sustainable synthesis of N-heterocycles in water using alcohols following the double dehydrogenation strategy. J. Catal. 2019, 373, 93-102.
40. Deshmukh, G.; Gharpure, S. J.; Murugavel, R. Dinuclear Ru(II) schiff base complex catalyzed one-pot synthesis of quinolines through acceptorless dehydrogenative coupling of secondary alcohols with 2-nitrobenzyl alcohol. Organometallics 2024, 43, 1190-202.
41. Wang, Q.; Wang, M.; Li, H.; Zhu, S.; Liu, Y.; Wu, Y. Synthesis of quinolines via iron-catalyzed redox condensation of alcohols with 2-nitrobenzyl methyl ether/2-nitrobenzyl alcohols. Synthesis 2016, 48, 3985-95.
42. Xi, J.; Jung, H. S.; Xu, Y.; Xiao, F.; Bae, J. W.; Wang, S. Synthesis strategies, catalytic applications, and performance regulation of single-atom catalysts. Adv. Funct. Mater. 2021, 31, 2008318.
43. Guo, W.; Wang, Z.; Wang, X.; Wu, Y. General design concept for single-atom catalysts toward heterogeneous catalysis. Adv. Mater. 2021, 33, e2004287.
44. Li, X.; Surkus, A. E.; Rabeah, J.; et al. Cobalt single-atom catalysts with high stability for selective dehydrogenation of formic acid. Angew. Chem. Int. Ed. 2020, 59, 15849-54.
45. Li, B.; Kou, J.; Zeng, G.; Ma, J.; Dong, Z. Atomically dispersed Co on N-doped porous carbon for the highly efficient catalytic oxidation of aromatic alcohols to acids and nitriles. ACS. Catal. 2023, 13, 16286-99.
46. Ping, K.; Bhadoria, R.; Starkov, P.; Kongi, N. M-N-C materials as heterogeneous catalysts for organic transformations. Coord. Chem. Rev. 2023, 497, 215412.
47. Sun, J. L.; Jiang, H.; Dixneuf, P. H.; Zhang, M. Reductive coupling of nitroarenes and HCHO for general synthesis of functional ethane-1,2-diamines by a cobalt single-atom catalyst. J. Am. Chem. Soc. 2023, 145, 17329-36.
48. Li, M.; Hu, W.; Wang, B.; et al. Mechanism of hydrogen generation catalyzed by a single atom and its spin regulation. J. Am. Chem. Soc. 2025, 147, 6193-202.
49. Levell, Z.; Yu, S.; Wang, R.; Liu, Y. What is the "other" site in M-N-C? J. Am. Chem. Soc. 2025, 147, 603-9.
50. Chen, S.; Luo, T.; Li, X.; et al. Identification of the highly active Co-N4 coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 2022, 144, 14505-16.
51. Li, W.; Meng, R.; Wang, K.; Cheng, Y.; Cai, D.; Zhan, G. Engineering pyridinic-N-Co sites for enhanced CO2 hydrogenation to methanol. Appl.Catal. B. Environ. Energy. 2025, 365, 124906.
52. Frisch, M.; Trucks, G.; Schlegel, H.; et al. Gaussian 16, Revision C.01. Wallingford, CT: Gaussian, Inc.; 2016. Available from: https://gaussian.com/citation/ [Last accessed on 28 May 2026].
53. Lu, T.; Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580-92.
54. Humphrey, W.; Dalke, A.; Schulten; K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33-8.
55. Liang, L.; Jin, H.; Zhou, H.; et al. Cobalt single atom site isolated Pt nanoparticles for efficient ORR and HER in acid media. Nano. Energy. 2021, 88, 106221.
56. Ha, Y.; Fei, B.; Yan, X.; et al. Atomically dispersed Co‐pyridinic N‐C for superior oxygen reduction reaction. Adv. Energy. Mater. 2020, 10, 2002592.
57. Chen, M.; Wang, N.; Zhu, L. Single-atom dispersed Co-N-C: a novel adsorption-catalysis bifunctional material for rapid removing bisphenol A. Catal. Today. 2020, 348, 187-93.
58. Zhang, G.; Lu, W.; Cao, F.; Xiao, Z.; Zheng, X. N-doped graphene coupled with Co nanoparticles as an efficient electrocatalyst for oxygen reduction in alkaline media. J. Power. Sources. 2016, 302, 114-25.
59. Yang, T.; Li, K.; Pu, L.; et al. Hollow-spherical Co/N-C nanoparticle as an efficient electrocatalyst used in air cathode microbial fuel cell. Biosens. Bioelectron. 2016, 86, 129-34.
60. Niu, K.; Yang, B.; Cui, J.; et al. Graphene-based non-noble-metal Co/N/C catalyst for oxygen reduction reaction in alkaline solution. J. Power. Sources. 2013, 243, 65-71.
61. Liu, X.; Gu, Q.; Zhang, Y.; et al. Atomically thick oxide overcoating stimulates low-temperature reactive metal-support interactions for enhanced catalysis. J. Am. Chem. Soc. 2023, 145, 6702-9.
62. Zhang, G.; Tang, F.; Wang, X.; Wang, L.; Liu, Y. Atomically dispersed Co-S-N active sites anchored on hierarchically porous carbon for efficient catalytic hydrogenation of nitro compounds. ACS. Catal. 2022, 12, 5786-94.
63. Lin, X.; Zhao, S.; Chen, Y.; Fu, L.; Zhu, R.; Liu, Z. Nitrogen-doped carbon cobalt grafted on graphitic carbon nitride catalysts with enhanced catalytic performance for ethylbenzene oxidation. J. Mol. Catal. A. Chem. 2016, 420, 11-7.
64. Zhu, Q.; Wang, F.; Zhang, F.; Dong, Z. Renewable chitosan-derived cobalt@N-doped porous carbon for efficient aerobic esterification of alcohols under air. Nanoscale 2019, 11, 17736-45.
65. Gao, J.; Feng, L.; Ma, R.; et al. Cobalt single-atom catalysts for domino reductive amination and amidation of levulinic acid and related molecules to N-heterocycles. Chem. Catal. 2022, 2, 178-94.
66. Wang, C.; Liu, Y.; Ren, H.; Guan, Q.; Chou, S.; Li, W. Diminishing the uncoordinated N species in Co-N-C catalysts toward highly efficient electrochemical CO2 reduction. ACS. Catal. 2022, 12, 2513-21.
67. Du, Z.; Chen, X.; Hu, W.; et al. Cobalt in nitrogen-doped graphene as single-atom catalyst for high-sulfur content lithium-sulfur batteries. J. Am. Chem. Soc. 2019, 141, 3977-85.
68. Lu, X.; He, J.; Huang, L.; et al. Synergetic roles of pyridinic nitrogen and carbonyl sites in nitrogen-doped carbon for the metal-free transfer hydrogenation reactions. Appl. Catal. B. Environ. 2023, 324, 122277.
69. Yang, H.; Cui, X.; Dai, X.; Deng, Y.; Shi, F. Carbon-catalysed reductive hydrogen atom transfer reactions. Nat. Commun. 2015, 6, 6478.
70. Luo, Z.; Nie, R.; Nguyen, V. T.; et al. Transition metal-like carbocatalyst. Nat. Commun. 2020, 11, 4091.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].