


Advanced materials and computational methods in potassium-sulfur batteries
 0
0 Abstract
Potassium-sulfur (K-S) batteries have emerged as an ideal candidate for large-scale energy storage because of the high theoretical energy density and low material cost. However, the practical deployment of K-S batteries is impeded by challenges such as polysulfide shuttling, sluggish conversion kinetics, and interfacial instability. Despite extensive experimental work in materials development, the underlying mechanisms at the atomic and molecular levels remain poorly understood. Computational methods have proven useful for elucidating K-S electrochemistry from a microscopic perspective. Nevertheless, a comprehensive review that systematically combines these experimental advances with computational insights is still lacking. To address this gap, this review provides a comprehensive overview of advances in K-S battery research, with emphasis on the materials engineering and computational research. We first summarize the fundamental mechanisms of K-S batteries and progress in key battery components including cathode, anode, electrolyte, as well as binder and separator. Complementing these experimental efforts, we introduce the theoretical foundations of density functional theory and molecular dynamics simulations, and review their recent applications in critical aspects such as adsorption energetics, reaction kinetics, electronic properties, and dynamic behaviors. Finally, we outline future directions for K-S battery research, including the integration of advanced characterization with multi-scale simulation, the development of high-throughput experimental and computational platforms, and the application of artificial intelligence to accelerate the development of materials and computational simulations. This review aims to provide guidance for the rational design of high-performance K-S battery systems by integrating experimental and theoretical insights.
Keywords
INTRODUCTION
To mitigate climate change and environmental pollution, the global energy landscape is rapidly transitioning toward clean and renewable sources[1,2]. However, the inherent intermittency and variability of green energy technologies such as wind and solar power constrain their reliable supply, underscoring the critical need for advanced energy storage systems[3-5]. Lithium-ion batteries (LIBs) are currently the most advanced power source for portable electronics and electric vehicles, and they also show great promise for grid-scale energy storage[6,7]. Nevertheless, constrained by the low theoretical specific capacities of cathode materials (e.g., layered oxides, 150-200 mAh g-1) and anode materials (e.g., graphite, 372 mAh g-1), the energy density of commercial LIBs is generally limited to no more than about 350 Wh kg-1, which falls short of meeting the growing global energy demand[8-11].
Based on sulfur conversion chemistry, lithium-sulfur (Li-S) batteries are an attractive alternative to conventional LIBs, capable of supporting fast-expanding energy demands[12]. Specifically, each sulfur atom in Li-S batteries undergoes a reversible two-electron redox reaction, enabling the sulfur cathode to deliver a remarkably high theoretical specific capacity of up to 1,675 mAh g-1, which is much higher than traditional transition-metal oxide cathodes[13]. Moreover, sulfur is naturally abundant and low-cost, which makes it appealing as a cathode material. However, the scarcity (approximately 0.0017 wt.% in the earth’s crust) and high cost (industrial-grade lithium carbonate: 100 USD kg-1) of lithium (Li) resources constrain the further development of Li-S batteries[14]. In comparison, potassium-sulfur (K-S) batteries offer compelling advantages in terms of resource sustainability, electrochemical performance, and environmental compatibility. First, potassium (K) exhibits significantly higher natural abundance (approximately 1.5 wt.% in the earth’s crust) and lower material cost (potassium carbonate: 3-5 USD kg-1) than Li, endowing K-S batteries with enhanced economic viability and resource sustainability[15,16]. Second, despite the larger ionic radius of K+ (1.38 Å) compared to Li+ (0.76 Å) and Na+ (1.02 Å), K+ possesses a smaller Stokes radius in carbonate-based electrolytes (3.6 Å for K+, 4.8 Å for Li+ and 4.6 Å for Na+) due to its weaker Lewis acidity[17,18]. This leads to higher ionic conductivity, higher ion mobility, and a higher transport number for K+, which directly benefits rate capability in K-S batteries. Third, K has a lower redox potential than Li and Na in carbonate-based electrolytes (-0.09, 0, and 0.23 V, respectively, vs. Li/Li+), which helps to achieve higher full-cell operating voltages[19]. Fourth, the widespread use of K in agricultural applications highlights its environmental compatibility, further strengthening the appeal of K-S batteries as a green energy storage technology[20].
From the battery system perspective, although the theoretical gravimetric energy density of K-S batteries (~1,023 Wh kg-1) is lower than that of Li-S batteries (~2,571 Wh kg-1) and sodium-sulfur (Na-S) batteries (~1,273 Wh kg-1), it still significantly exceeds that of commercial LIBs [Table 1][21]. Moreover, K-S batteries use low-cost aluminum instead of copper as the anode current collector, further reducing manufacturing costs compared to Na-S batteries. Nevertheless, K-S batteries also face distinct challenges that are more severe than those in Li-S and Na-S systems. The large size of K+ causes dramatic volume expansion (296% for S to K2S, 80% for Li2S, 170% for Na2S) during cycling, leading to rapid electrode pulverization and capacity fading[8]. Moreover, the solid electrolyte interphase (SEI) formed on K metal anodes, mainly composed of K2CO3, K2O, and KF, is more soluble and less stable than the Li or Na counterparts, resulting in persistent side reactions and dendrite growth[22]. The dissolution and shuttling of potassium polysulfides, although analogous to the Li-S system, may exhibit different thermodynamics and binding affinities with host materials, an area that remains poorly understood[15].
Comparisons of Li-S, Na-S, and K-S batteries
| Battery types | Li-S batteries | Na-S batteries | K-S batteries |
| Conversion mechanism | 2Li + 1/8S8 ⇋ Li2S | 2Na + 1/8S8 ⇋ Na2S | 2K + 1/8S8 ⇋ K2S |
| Standard reaction potential (vs. SHE, V) | 2.29 | 1.81 | 1.89 |
| Energy density (Wh kg-1) | 2,571 | 1,273 | 1,023 |
| Volume expansion upon sulfur conversion | 80% | 170% | 296% |
| Reaction kinetics | Fast | Slow | Medium |
| Cost | High | Low | Medium |
Undoubtedly, research on K-S batteries has produced a large number of encouraging results since their first report in 2014 [Figure 1A]. Significant advances have been made in key components such as cathodes, anodes, electrolytes, binders, and separators [Figure 1B and Table 2][23-43]. Despite these achievements, the lack of a deep understanding of the underlying mechanisms will hinder the rapid long-term development of K-S batteries. For example, carbon hosts have been experimentally shown to effectively suppress polysulfide dissolution, but it remains difficult to unveil how the pore structure and surface chemistry of carbon materials influence the immobilization and conversion of potassium polysulfides at the atomic level. In recent years, advanced computational methods have become indispensable tools for elucidating the complex chemistry of K-S batteries and accelerating materials development[44,45]. Density functional theory (DFT) calculations can accurately analyze electronic structures, bonding interactions, and reaction pathways at the atomic scale[46], making them particularly suitable for revealing how host materials anchor polysulfides and catalyze their conversion. Molecular dynamics (MD) simulations can reveal dynamic behavior, structural evolution, and thermodynamic properties over extended time scales[47], offering unique insights into ion transport in electrolytes, polysulfide dissolution behavior, and the formation and evolution of the SEI. These advanced computational methods are expected to elucidate the core mechanism of K-S electrochemistry, thereby guiding the rational design of high-performance systems.
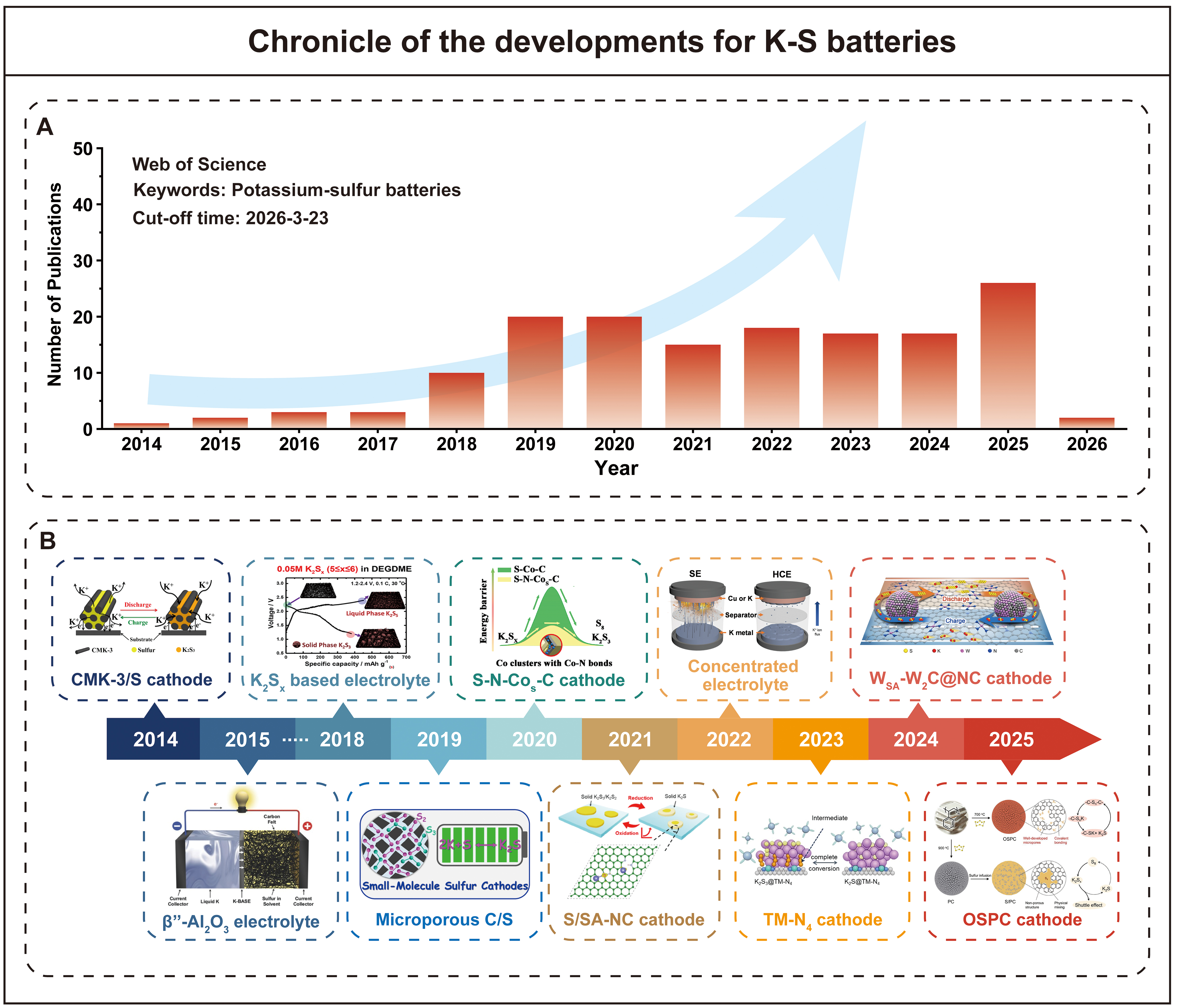
Figure 1. Chronicle of the developments for K-S batteries. (A) The number of papers published each year relevant to “Potassium-sulfur batteries” from 2014 to 2026. (B) Significant research achievements in the development of K-S batteries (Reproduced with permission from[23]. Copyright 2014, American Chemical Society Reproduced with permission from[24]. Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Reproduced with permission from[25]. Copyright 2018, American Chemical Society Reproduced with permission from[26]. Copyright 2019, American Chemical Society Reproduced with permission from[27]. Copyright 2020, American Chemical Society Reproduced with permission from[28]. Copyright 2021, American Chemical Society Reproduced with permission from[29]. Copyright 2022, Wiley-VCH GmbH Reproduced with permission from[30]. Copyright 2023, Wiley-VCH GmbH Reproduced with permission from[31]. Copyright 2024, Springer Nature Reproduced with permission from [32]. Copyright 2025, Wiley-VCH GmbH).
Electrochemical performances of K-S batteries with representative materials
| Electrode | Electrolyte | Separator | Working current | Cycle number | Retained discharge capacity | Ref. |
| PANI@CMK-3/S||K | 1.0 M KClO4 in TEGDME | Glass fiber | 50 mA g-1 | 50 | 329.3 mAh g-1 | [23] |
| Microporous C/S||K | 0.8 m KPF6 in EC/DEC | Celgard2400 | 20 mA g-1 | 150 | 869.9 mAh g-1 | [26] |
| CNF/S||K | 1.0 M KCF3SO3 in TEGDME | Celgard/SWCNT | C/10 | 50 | ~600 mAh g-1 | [33] |
| SPAN||K | 0.8 m KPF6 in EC/DEC | Glass fiber | 0.5 C | 100 | 147 mAh g-1 | [34] |
| SPAN with PAA binder||K | 0.5 m KPF6 in EC/DMC | Glass fiber | 837.5 mA g-1 | 100 | ~998 mAh g-1 | [35] |
| CCS||K | 1.0 M KCF3SO3 in EC/DEC | Glass fiber | 150 mA g-1 | 300 | 253 mAh g-1 | [36] |
| I-S@pPAN||K | 0.8 m KPF6 in EC/DEC | Glass fiber | 0.1 C | 100 | 722 mAh g-1 | [37] |
| K2Sx (5 ≤ x ≤ 6) catholyte + 3D-FCN||hard carbon | 0.05 M K2Sx (5 ≤ x ≤ 6) in DEGDME | Glass fiber + Celgard2400 | 0.1 C | 1 | 235 mAh g-1 | [25] |
| SPAN||liquid K-Na | 0.8 m KPF6 in EC/DMC/EMC | Glass fiber | 35 mA g-1 | 100 | 490 mAh g-1 | [38] |
| CMK-3/S||K | 5 M KTFSI in DEGDME | Glass fiber | 10 mA g-1 | 10 | / | [39] |
| S + K2Sx in 1.5 M KTFSI in tetraglyme||K | K-β"-Al2O3 | / | C/4.2 | 1,000 | / | [24] |
| SeS2@NCNFs||K | 0.7 m KPF6 in EC/DEC | Glass fiber | 0.5 A g-1 | 1,000 | 417 mAh g-1 | [40] |
| Ga-Cd DAs-HMCS/S||K | 3 M KTFSI in EC | Glass fiber | 1 A g-1 | 500 | 649 mAh g-1 | [41] |
| SPAN||KC8 | KFSI/TMP/HFE = 1:8.7:8.7, molar ratio | Glass fiber | 50 mA g-1 | 50 | / | [42] |
| NC/S||K | 0.8 m KPF6 in EC/DEC | GF@SnSe | 0.2 C | 180 | 436 mAh g-1 | [43] |
This review provides a systematic overview of advances in materials research for K-S batteries, while highlighting the application of advanced computational simulation techniques in this emerging field. We begin by summarizing the fundamental mechanisms and the progress made in key components of K-S batteries including cathode, anode, electrolyte, as well as binder and separator. Subsequently, we introduce the theoretical foundations of widely used computational methods (DFT and MD methods), and review their recent applications in critical aspects such as adsorption energetics, reaction kinetics, electronic properties, and dynamic behaviors. Finally, we offer perspectives on future directions for K-S battery research.
ADVANCED MATERIALS ENGINEERING IN K-S BATTERIES
Fundamental mechanisms of K-S batteries
Before delving into the material design, it is essential to fully understand the underlying electrochemical mechanisms of K-S batteries. A typical K-S battery consists of K metal as the anode and sulfur as the cathode. During discharge, K metal is oxidized to K+ and releases electrons that travel through the external circuit, while sulfur at the cathode is gradually reduced through a series of potassium polysulfide intermediates[8]. The specific electrochemical reactions are shown as follows[17]:
On the cathode side, the reduction pathway strongly depends on the initial allotrope of sulfur[21]. For cyclo-octasulfur, the reduction first proceeds to long-chain polysulfides (K2Sn, 5 ≤ n ≤ 8), then further to short-chain polysulfides (K2Sn, 2 ≤ n ≤ 4), and finally to potassium sulfide (K2S). In contrast, for low-order chain-like sulfur allotropes, the reduction bypasses the formation of long-chain polysulfides and converts more directly to the final product.
In the actual operation of batteries, the reaction sequence of potassium polysulfide intermediates is not fully understood, which is a key issue worthy of further exploration. One of the most distinctive features of the K-S system is the existence of multiple stable K2Sn phases at room temperature, including n = 1, 2, 3, 4, 5, 6. This stands in stark contrast to the Li-S system, which has only one equilibrium phase (Li2S)[15]. Each two-phase region (K2Sn and K2Sn+1) would theoretically produce a plateau in the discharge curve. However, due to reaction overpotentials and resistive losses, these plateaus merge into a sloping curve[15]. The discharge voltage profile of K-S batteries is qualitatively similar to that of Li-S systems, but with less distinct plateaus, exhibiting a sloping shape, lower overall redox voltages, and larger charge-discharge hysteresis[21]. Therefore, it is not clear whether the K-S system and the Li-S system have the same sequence of polysulfide intermediates. More importantly, even the final discharge product of K-S batteries is still under debate. In early work using mesoporous carbon (CMK-3) as the sulfur host, X-ray diffraction analysis revealed that the potassiation product after discharge to 1.2 V was K2S3, and it was argued that further reduction of K2S3 to K2S is thermodynamically unfavorable[23]. However, later studies using small-molecule sulfur confined in microporous carbon or covalently bonded sulfur achieved reversible capacities exceeding the theoretical limit of K2S3, and provided direct experimental evidence for the formation of K2S[26,34,35]. These findings suggest that the difficulty in achieving the conversion from K2S3 to K2S is likely kinetic rather than thermodynamic in nature. Theoretical calculations offer additional insights into the relative stability of these phases. Among various potassium sulfides, K2S is the most thermodynamically stable phase with the lowest formation energy, while K2S2 has a higher formation energy than both K2S3 and K2S, making it prone to disproportionation into K2S3 and K2S[8].
Cathode
Carbon based composite cathode
Similar to Li-S batteries and Na-S batteries, carbon materials have also been studied as sulfur host materials for K-S batteries[8]. The porous structure of carbon materials can not only confine active sulfur species and adsorb potassium polysulfide to suppress the shuttle effect, but also alleviate volume expansion. The high specific surface area of carbon materials is conducive to electrolyte infiltration and ion transport. More importantly, carbon materials typically have good conductivity.
Among various carbon materials, mesoporous carbon is the most widely used in Li-S and Na-S battery systems, which has also been extended to K-S batteries[23]. Zhao et al.[23] from Nankai University successfully constructed a rechargeable K-S battery for the first time using ordered mesoporous carbon CMK-3 as the carrier of sulfur, as shown in Figure 2A. They prepared CMK-3/sulfur composite cathode materials by the melt diffusion method. To further improve battery performance, they also applied the in-situ polymerization method to coat a conductive polyaniline (PANI) layer on the surface of the composite material. Through characterization analysis, they confirmed that the discharge process mainly generates K2S3, which reversibly decomposes into sulfur and K+ during charging. Electrochemical tests showed that the CMK-3/sulfur composite material had a first cycle discharge capacity of 512.7 mAh g-1 at a current density of 50 mA g-1, and remained at 202.3 mAh g-1 after 50 cycles[23]. After introducing polyaniline coating, the cycling performance of the optimized battery was further enhanced, which delivered a capacity of 329.3 mAh g-1 after 50 cycles, while the sulfur loss rate decreased from 62% to 28%.
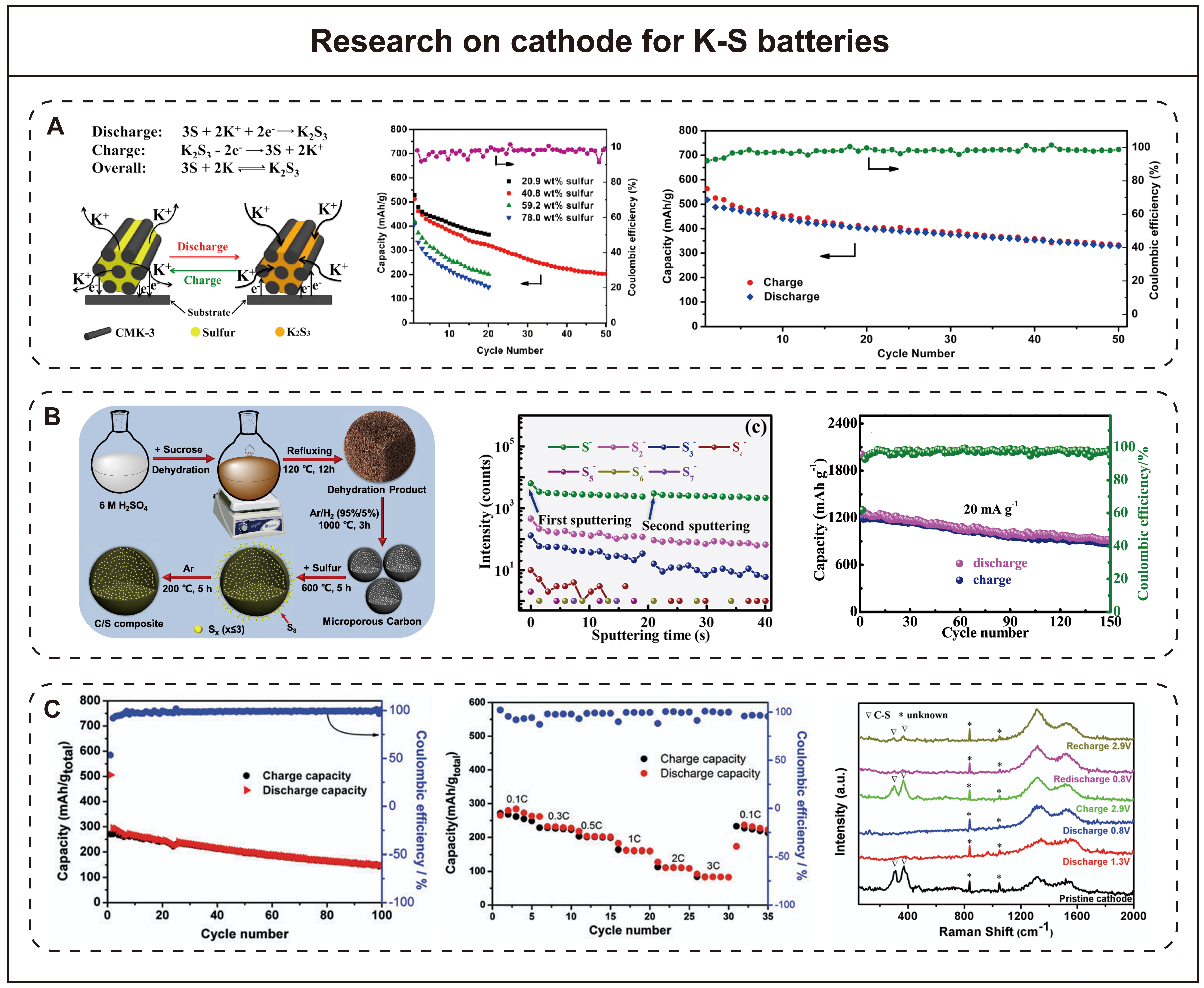
Figure 2. Research on cathode for K-S batteries. (A) PANI@CMK-3/sulfur composite cathode (Reproduced with permission from[23]. Copyright 2014, American Chemical Society). (B) Microporous carbon-confined small-molecule sulfur cathode (Reproduced with permission from[26]. Copyright 2019, American Chemical Society). (C) SPAN cathode (Reproduced with permission from[34]. Copyright 2018, The Royal Society of Chemistry).
However, polysulfides in mesoporous carbon pores may escape during battery cycling. To avoid the shuttle effect, soluble polysulfides must either be prevented from forming or completely restricted by the host material. These two issues can be solved by placing small-molecule sulfur in microporous carbon to directly generate insoluble short-chain polysulfides in the electrochemical reaction, or by introducing specific species or functional groups to immobilize polysulfides and rapidly convert them. Xiong et al.[26] designed a composite cathode material using microporous carbon as a support and loading small-molecule sulfur for room-temperature K-S batteries [Figure 2B]. They confined sulfur in the microporous carbon matrix through high-temperature gas-phase treatment and stabilized sulfur into small molecules through the strong interaction between microporous carbon and sulfur. According to time-of-flight secondary ion mass spectrometry results, they confirmed that there was no high molecular weight sulfur above S8 in the microporous carbon, thus eliminating the formation of soluble polysulfides. Other characterization experiments also showed that the composite material has excellent resistance to sulfur volatilization and decomposition[26]. As a result, the cathode has a capacity of 1,198.3 mAh g-1 at 20 mA g-1 and still maintains 869.9 mAh g-1 after 150 cycles, with a Coulombic efficiency of 97%. Combining X-ray photoelectron spectroscopy analysis and theoretical calculations, they further clarified that the resulting product is K2S, with a theoretical capacity of 1,675 mAh g-1[26]. To achieve the immobilization and rapid conversion of soluble polysulfides, Ge et al.[27] designed a nitrogen-doped cobalt nanoclusters/porous nitrogen-doped carbon composite material and used it as the cathode catalytic host for K-S batteries. They employed a low-temperature two-step pyrolysis strategy, preserving nitrogen doping and the porous structure, and through the strong interaction between sulfur and cobalt, fragmented cobalt particles into ultra-small clusters, significantly increasing the exposed catalytic active sites. They demonstrated that these cobalt nanoclusters and cobalt-nitrogen bonds can accelerate the conversion of polysulfides through electrochemical testing and theoretical calculations. This cathode maintained a capacity of 453 mAh g-1 after 50 cycles at 50 mA g-1. Even at 400 mA g-1, it still provided a high capacity of 415 mAh g-1. Furthermore, the stepwise transformation pathway S8 → K2S6 → K2S5 → K2S4 → K2S3 was revealed. Thermodynamic calculations also support the spontaneity of these intermediate steps.
Sulfur has poor electrical conductivity, which limits its utilization. A simple approach is to partially replace sulfur with other elements from the same group[15]. For example, selenium (Se) has much better electrical conductivity than sulfur[48]. Furthermore, Se undergoes a single-step conversion pathway during charging and discharging, avoiding the formation of soluble polyselenides. This prevents the shuttle effect. In addition, selenium is compatible with carbonate electrolytes. Yao et al.[40] prepared a self-supporting electrode material by confining SeS2 within the nitrogen-doped porous carbon nanofiber film. Electrochemical analysis showed that during the cathode potassiation process, SeS2 reacts with K to generate K2Se and K2S. During depotassiation, K2Se and K2S are directly converted to SeS2[40]. Furthermore, the three-dimensional interconnected porous carbon network of this composite material provides a rapid transport channel for electrons and K+ and effectively buffers volume changes during charge and discharge. Theoretical calculations confirmed that the high content of pyrrole nitrogen and pyridine nitrogen in the carbon fibers significantly enhances the chemisorption capacity for discharge products, thereby effectively suppressing the shuttle effect of polysulfides and polyselenides. This electrode exhibits excellent electrochemical performance in carbonate electrolytes. After 150 cycles at 0.05 A g-1, it still maintains a high reversible capacity of
Covalent polymer-sulfur cathode
The idea behind covalent polymer sulfur cathodes is to bind sulfur to a conductive framework via covalent bonds, ensuring that the sulfur remains bonded to the carrier throughout battery cycling. Sulfurized polyacrylonitrile (SPAN) is a classic example of this type of material. In 2018, Liu et al.[34] first applied SPAN to a room-temperature K-S battery and systematically studied its potassium storage behavior in a carbonate electrolyte [Figure 2C]. They successfully prepared the SPAN nanocomposite by treating polyacrylonitrile and sulfur at high temperature under an argon atmosphere using a one-step solid-state method. Raman spectroscopy, infrared spectroscopy, and X-ray diffraction analysis confirmed that the sulfur existed in a small-molecule form and was linked to the carbon matrix via C-S covalent bonds. In electrochemical tests, the performance of SPAN was evaluated using K as the anode and 0.8 M potassium hexafluorophosphate dissolved in ethylene carbonate (EC)/diethyl carbonate (DEC) as the electrolyte[34]. The initial reversible capacity of SPAN was 270 mAh g-1 at 0.5 C, and it exhibited excellent rate performance in the range of 0.1 C to 3 C. To investigate the reaction mechanism, the team detected the Raman signal of the SPAN electrode during the battery cycling process. The results showed that the C-S bond characteristic peak disappeared after discharge and reappeared after charging. As the number of cycles increased, the intensity of the C-S bond peak gradually weakened, indicating that the process was not completely reversible. X-ray photoelectron spectroscopy analysis also supported this conclusion[34]. Upon charging the battery to 2.9 V, a small amount of sulfate/thiosulfate species were detected, which is due to some discharge products not being completely converted back to their original state.
To further improve electrode performance, Ma et al.[37] introduced iodine doping into the SPAN to obtain an I-S@pPAN composite material. Characterization results showed that iodine was uniformly doped into the framework in the form of C-I covalent bonds. The interlayer distance of the carbon layers was increased by introducing iodine. The performance of the iodine-doped SPAN in K-S cells was significantly better than that of the original SPAN. At a current density of 0.1 C, the discharge capacity of I-S@pPAN reached
Anode
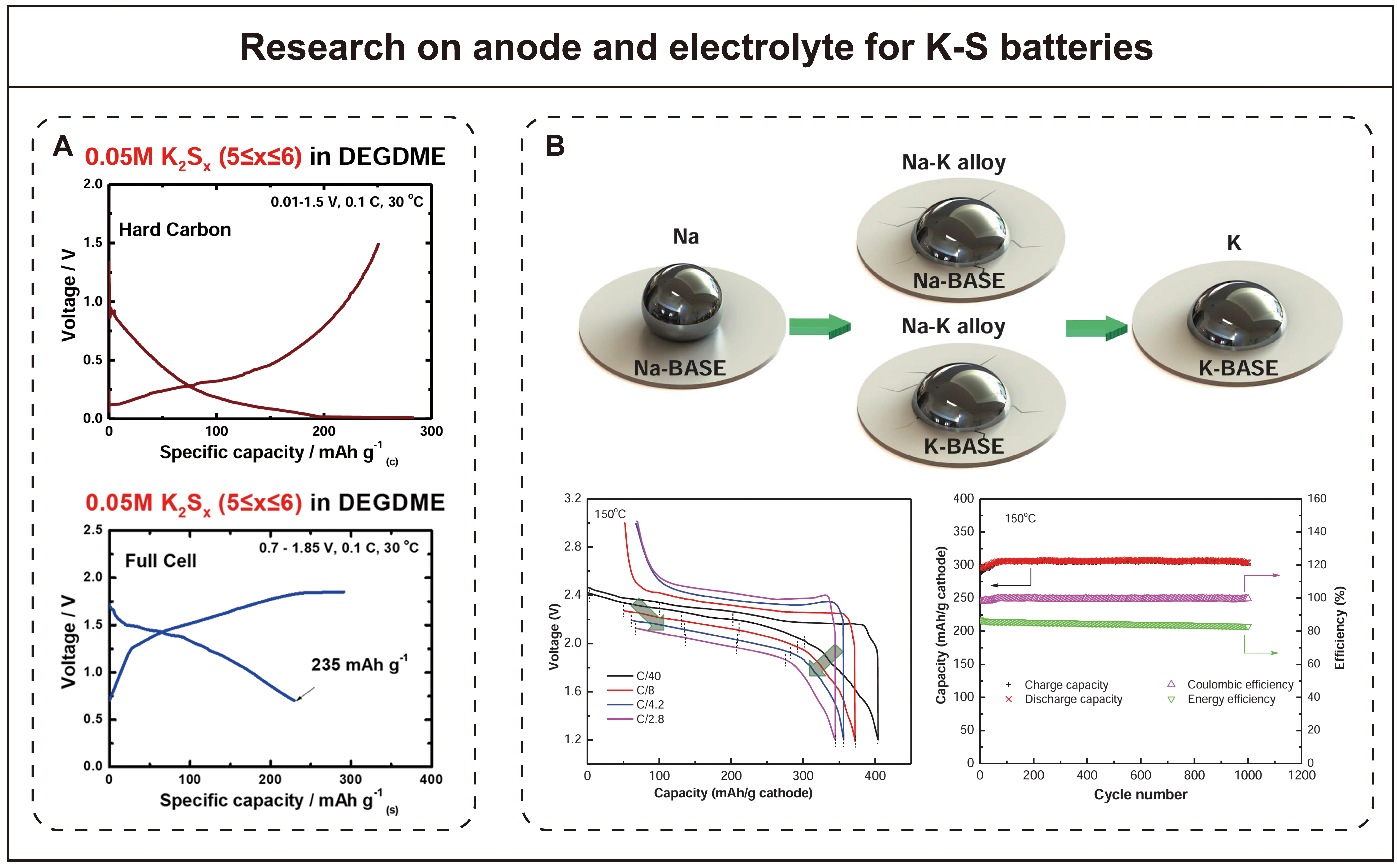
Developing a stable and safe anode is a major challenge for K-S batteries. Although K metal provides a high theoretical specific capacity of 687 mAh g-1, its high reactivity makes it easy to react with electrolytes and sulfides. The associated unstable K deposition/stripping behavior and dendrite growth problems are also not to be underestimated[49]. Interestingly, unlike Li or Na, K dendrites tend to exhibit a moss-like morphology and preferentially grow at the root instead of the tip, a unique characteristic that is still poorly understood[50,51]. In addition, the SEI formed on potassium anodes, composed of species such as K2CO3, K2O, and KF, is often unstable and leads to heterogeneous ion flux, further exacerbating dendrite formation[22]. To address these issues, anode designs have been explored. Zhang et al.[38] proposed using a liquid K-Na alloy as the anode in K-S batteries, instead of K metal. This alloy contains between 41.8% and 90.8% K and remains liquid at room temperature. Because the anode is liquid, K dendrites will not grow during charge and discharge, which improves battery safety to some extent. The research team injected the liquid alloy into specially-made carbon fiber paper and then matched it with a SPAN cathode to assemble a novel K-S battery. Electrochemical tests showed that this battery with the K-Na alloy anode maintained a capacity of
Electrolyte
Electrolyte is considered as a core component of a battery, and its impact on battery performance is decisive. Current optimization efforts for K-S battery electrolytes mainly focus on suppressing the shuttle effect of polysulfides. For liquid electrolytes, increasing the electrolyte concentration is a very effective approach. High-concentration electrolytes typically contain fewer free solvent molecules, which directly reduces the solubility of polysulfides, compared to conventional electrolytes. The higher viscosity of high-concentration electrolytes also hinders the diffusion of polysulfides[15,17]. Furthermore, high-concentration electrolytes are compatible with K metal, which can form a stable SEI, suppressing interfacial side reactions and dendrite growth[51]. Wang et al.[39] explored the effect of electrolyte concentration on the electrochemical behavior of K-S batteries. Electrochemical tests showed that batteries filled with low-concentration electrolyte (1 M) could only provide an initial discharge capacity of 285 mAh g-1 at 10mA g-1[39]. However, when the electrolyte concentration was increased to 5 M, the battery achieved an initial discharge capacity of 606 mAh g-1, while the charge capacity reached 527 mAh g-1, indicating that polysulfide side reactions were effectively suppressed. Furthermore, the research team conducted visualization experiments[39]. The solution turned brown during discharge in 1 M and 3 M electrolytes, while the 5 M electrolyte was only light yellow, indicating that high-concentration electrolytes effectively suppressed polysulfide dissolution. Solid electrolytes have also been studied for blocking the shuttle effect of polysulfides. Lu and colleagues developed a β′′-Al2O3 solid electrolyte (K-BASE) for K-S battery [Figure 3B][24]. This K-BASE exhibits ionic conductivities of 0.056 S cm-1 at 300 °C and 0.01 S cm-1 at 150 °C. Simultaneously, liquid K exhibits excellent wettability on the K-BASE surface compared to the Na-based system, attributed to low surface tension of K and stronger adhesion work of K to solid electrolyte surface. This excellent wettability enables the K-S battery to operate at temperatures as low as 150 °C. The battery demonstrates good performance because the solid electrolyte blocks the polysulfide shuttle and side reactions. The K-S battery using K-BASE achieves a discharge capacity of 402 mAh g-1 at C/40. Furthermore, the battery also shows almost no capacity decay after 1,000 cycles at C/4.2. Shao et al.[52] reported a K+ conductor, K3SbS4, for room-temperature solid-state K-S batteries. The research team synthesized a series of K3-xSb1-xWxS4 compounds with high conductivity by doping the electrolyte with tungsten to increase the K vacancy concentration. Among them, the K2.92Sb0.92W0.08S4 material achieved an ionic conductivity of 7.7 × 10-5 S cm-1 at
Binder and separator
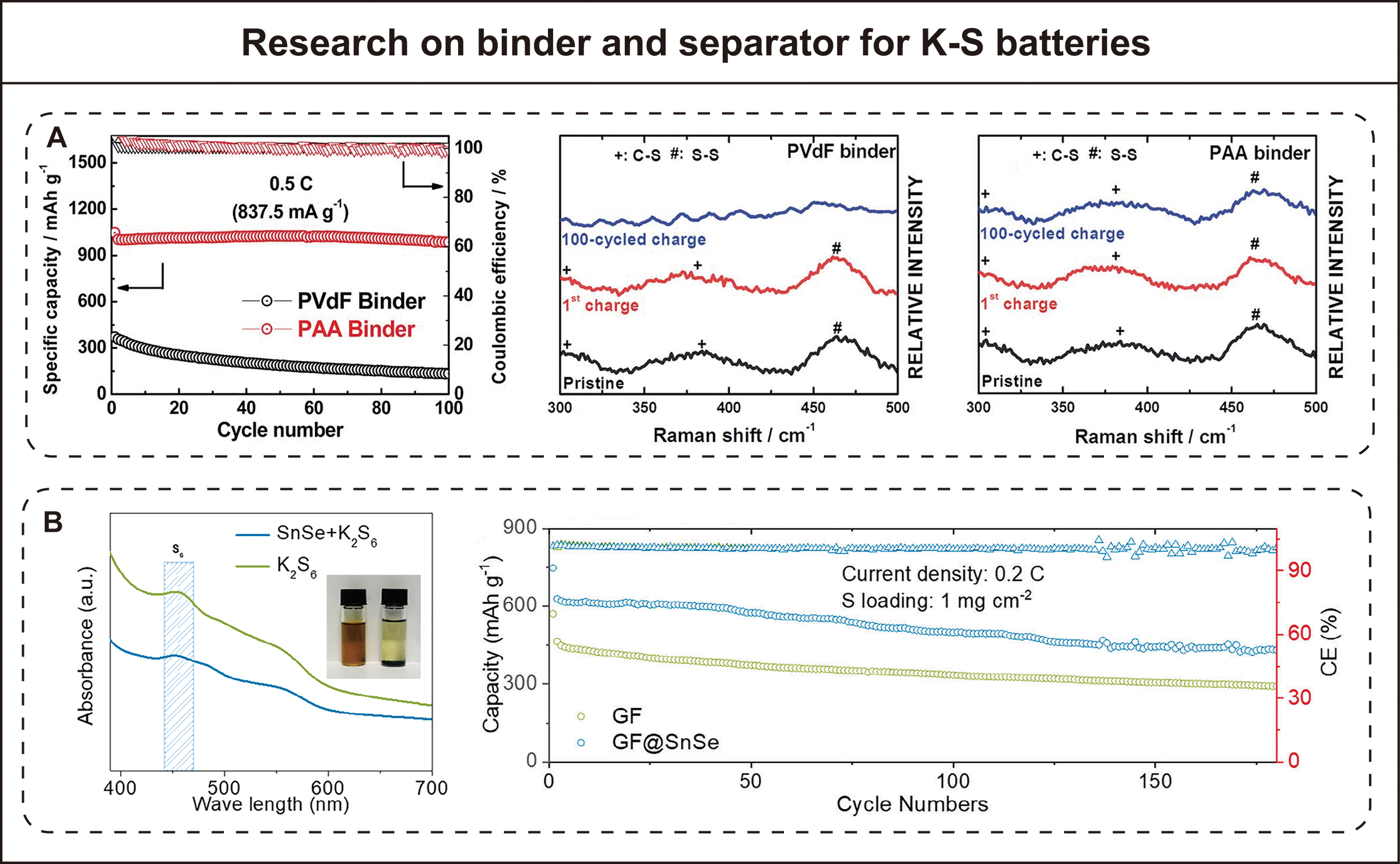
In addition to the crucial electrodes and electrolytes, some interesting research has also focused on binders and separators to optimize K-S battery performance. Regarding binders in K-S batteries, Sun’s team compared the performance of polyacrylic acid (PAA) and traditional polyvinylidene fluoride (PVdF) binders in SPAN cathodes [Figure 4A][35]. They found that PAA binder improved electrode uniformity and adhesion strength. This can cope with the drastic volume changes in the electrode caused by the large ionic radius K+ insertion/extraction. Through cyclic voltammetry testing, the SPAN electrode using PAA binder maintained highly reversible redox peaks after the first cycle, while the peak of the PVdF binder electrode decreased significantly, indicating that an irreversible reaction occurred. After 100 cycles at 0.5 C, the battery using PAA binder retained 95% of its capacity, but the battery using PVdF retained only 22%. Furthermore, the research team used Raman spectroscopy to analyze the electrode structure before and after cycling, confirming that PAA binder maintains the integrity of C-S bonds in SPAN[35]. Scanning electron microscopy and transmission electron microscopy observations further verified that PAA binder can maintain the particle structure and interfacial contact in the electrode. To address the rapid capacity decay caused by polysulfide shuttle, Manthiram’s group functionalized the commercial Celgard separator[33]. They used a vacuum filtration method to coat the surface of the Celgard separator with a layer composed of single-walled carbon nanotubes (SWCNTs). This three-dimensional nanonetwork structure of the SWCNT coating physically blocks the migration of soluble polysulfides. Furthermore, the good conductivity of SWCNTs allows them to act as an additional current collector, enabling the intercepted polysulfides to continue participating in electrochemical reactions at the separator-cathode interface. Performance tests showed that the K-S battery with SWCNT-coated separator achieved an initial discharge specific capacity of
ADVANCED COMPUTATIONAL METHODS IN K-S BATTERIES
Many key processes in K-S batteries, including polysulfide adsorption, ion diffusion, and the stability of discharge products, are essentially controlled by atomic and molecular interactions. Experimental techniques often face limitations in directly observing these nanoscale phenomena and elucidating their fundamental principles. To address this issue, computational methods have become indispensable tools, providing atomic-scale insights. Therefore, this section introduces the fundamental theory of widely used computational methods (DFT and MD methods) and their specific applications in K-S battery research.
DFT calculations
Theoretical foundations of DFT
DFT is based on quantum mechanics[53]. The stationary Schrödinger equation describes microscopic particles[54]:
where
In 1964, Hohenberg and Kohn proposed the Hohenberg-Kohn theorem, which states that the electron density distribution ρ(
In 1965, Kohn and Sham proposed the Kohn-Sham equations, as shown below[57],
The difficulty in solving the Kohn-Sham equations lies in the fact that the specific form of the exchange-correlation functional VXC is unknown. Therefore, it is necessary to approximate its parameterized empirical form by fitting the energies and charge density distributions of systems that have been solved exactly. Numerous approximation methods have been developed. The local density approximation (LDA) is the most basic type of exchange-correlation functional, which depends only on the electron density at each point[58]. LDA performs well in systems where the electron distribution is uniform or varies slowly. However, in practical systems where electrons are strongly localized, the electron density can vary significantly, and the accuracy of LDA calculations drops considerably. The generalized gradient approximation (GGA) improves upon LDA by incorporating information about the gradient of the electron density, thus providing a better description of regions with large density variations[59]. With the development of computational methods, several variants of GGA have emerged, among which the Perdew-Burke-Ernzerhof (PBE)[60] and Perdew-Wang[61] functionals are the most frequently used. To further improve the accuracy of exchange-correlation functionals, researchers have developed hybrid functionals that linearly combine traditional LDA or GGA with the exact exchange term from Hartree-Fock theory[62]. The proportion of each component can be adjusted by changing the linear combination coefficients, allowing computational results to better match experimental data. Many hybrid functionals have been developed, such as HSE06[63], B3LYP[64]. Among the functionals mentioned above, the combination of the PBE functional with the projector augmented wave pseudopotential is a popular choice for simulating cathode materials, as it achieves a good balance between efficiency and accuracy[65]. The B3LYP hybrid functional has been used to optimize the geometry of battery materials[66], while the HSE06 hybrid functional is more suitable for calculating the band structures of sulfur-based matrices[41].
Applications of DFT for K-S batteries
DFT calculation is a practical tool for studying the fundamental physicochemical processes behind the performance of K-S batteries. It provides atomic-scale insights into interactions, energy changes, and reaction pathways, offering a theoretical foundation for understanding and designing battery components. Next, we will specifically introduce several applications of DFT calculations in K-S battery research, focusing on adsorption mechanisms, ion diffusion behavior, electronic structure properties, and reaction kinetics.
A key challenge in K-S batteries is the dissolution and shuttle of polysulfides[46]. Therefore, it is essential to effectively confine these substances within the cathode region. This confinement can typically be demonstrated through adsorption interactions between the polysulfides and the host material, which can be achieved by DFT calculations. For instance, Liang et al.[65] studied the adsorption behavior of polysulfides on a nitrogen-rich microporous carbon matrix by DFT methods [Figure 5A]. They constructed two carbon models, including a microporous nitrogen-doped carbon model to simulate the effects of pore engineering, and a non-porous graphene model as a reference. By calculating the binding energies of polysulfides on these surfaces, they demonstrated that the microporous carbon model exhibited more negative adsorption energies compared to the non-porous model, indicating stronger interactions. This enhanced adsorption is due to the combined action of microporous confinement and nitrogen doping, which together improve the chemical anchoring of polysulfides and suppress the shuttle effect in K-S batteries.
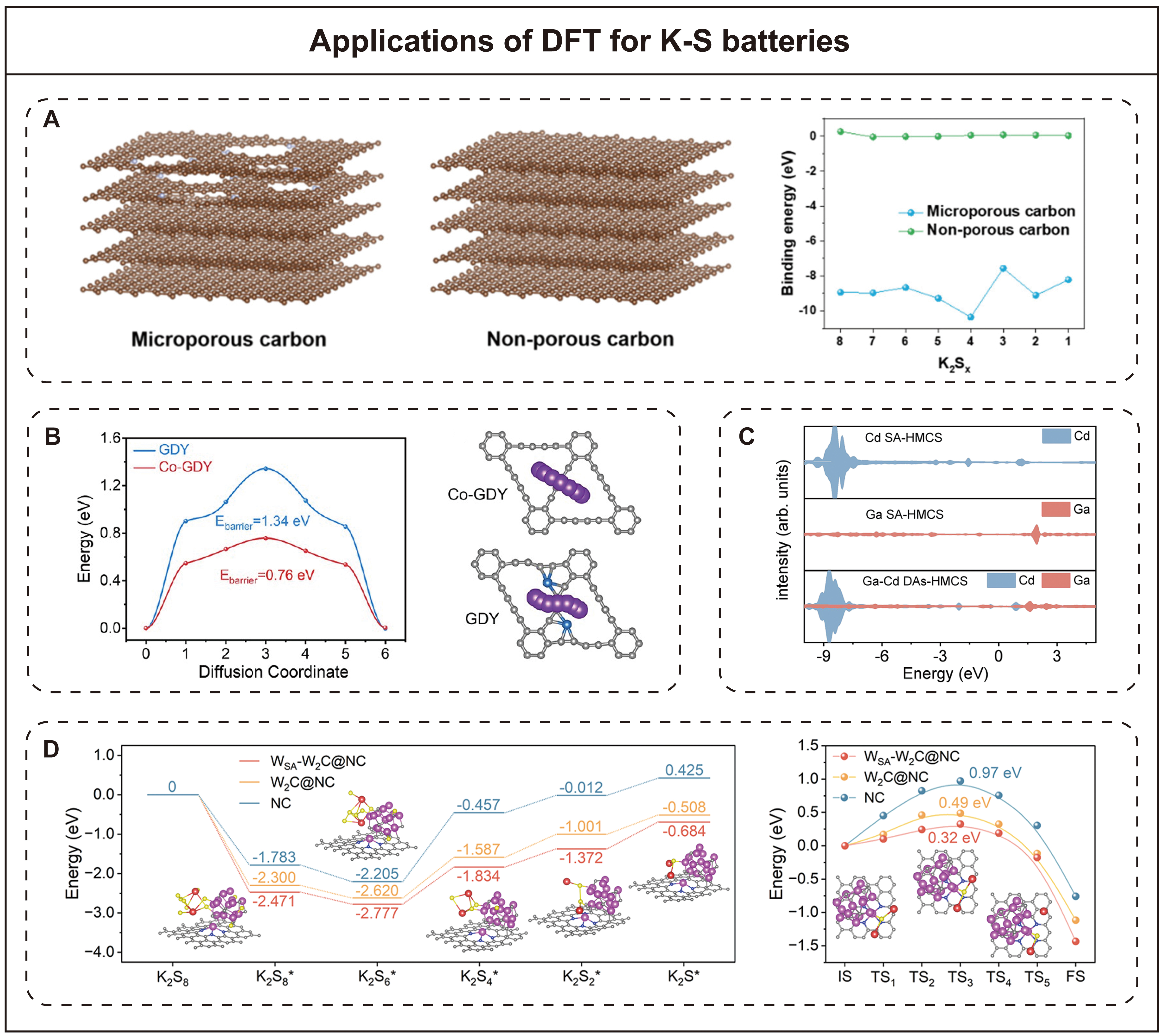
Figure 5. Applications of DFT for K-S batteries. (A) Adsorption interactions between polysulfides and the host material (Reproduced with permission from[65]. Copyright 2022, Multidisciplinary Digital Publishing Institute). (B) K+ diffusion behavior on the cathode (Reproduced with permission from[68]. Copyright 2024, American Chemical Society). (C) Electronic structure properties of cathode (Reproduced with permission from[41]. Copyright 2025, Springer Nature). (D) Reaction kinetics of conversion between polysulfide species (Reproduced with permission from[31]. Copyright 2024, Springer Nature).
Designing host materials with strong anchoring capabilities can enhance the adsorption of polysulfides, but most of them have poor conductivity, which usually leads to the accumulation of polysulfide species in electrochemically inactive regions[8]. In this case, the diffusion of polysulfide intermediates on the host material becomes a key factor affecting performance[67]. However, the diffusion of polysulfide species is usually inversely proportional to their adsorption strength on the host material, which makes it challenging to optimize both properties simultaneously. Diffusion simulations constructed by the DFT method can solve this complex problem. Zhang et al.[68] studied the energy barrier of K+ migration on the graphdiyne (GDY) and cobalt single-atom modified graphdiyne (Co-GDY), as shown in Figure 5B. Their results showed that the K+ migration barrier on Co-GDY (0.76 eV) was notably lower than that on GDY (1.34 eV), suggesting that the introduction of Co-C4 atomic sites with strong d-π orbital coupling effectively promoted K+ surface diffusion. This enhanced fluidity facilitates faster charge transfer and more efficient redox kinetics, thus contributing to improved performance of K-S batteries. The battery with Co-GDY maintained a discharge capacity of 1,064 mAh g-1 after 180 cycles at 0.2 A g-1, and its Coulombic efficiency was close to 100%.
The interaction between polysulfides and the host material depends on their electronic structure[44]. Electronic structure models have been widely used to elucidate the origins of different adsorption strengths and catalytic activities in metal-sulfur batteries. Transitions of electronic states to the Fermi level generally enhance electron transfer between the host and polysulfide species, thereby promoting anchoring ability and redox kinetics. For example, Zhang et al.[41] systematically investigated the electronic structures of Ga-Cd catalysts (DACs) immobilized on hollow mesoporous carbon spheres (Ga-Cd DAs-HMCS), which is shown in Figure 5C. By comparing the density of states of Ga-Cd DAs-HMCS and other single-atom counterparts, they found that adding Cd with fully filled valence electrons can transfer electrons to empty orbitals of Ga through strong orbital electron coupling[41]. This electronic interaction significantly increases the electron density of Ga sites near the Fermi level, thereby enhancing their catalytic activity for sulfur redox reactions.
The slow conversion between polysulfide species, especially the solid-state conversion to the final discharge product, limits the rate capability and cycle stability of the battery. The reaction kinetics controlling the interconversion of polysulfides can be studied by calculating the Gibbs free energy of intermediate sulfur species[44,46]. Smaller Gibbs free energy differences correspond to lower energy barriers and more favorable conversions. The climbing image nudged elastic band (CI-NEB) method is employed to assess the kinetic barriers of key reaction processes. Song et al.[31] investigated the sulfur reduction pathway and the decomposition behavior of K2S in K-S batteries [Figure 5D]. By calculating the Gibbs free energy for the stepwise reduction of sulfur on various host materials, they identified the conversion from K2S6 to K2S4 as the rate-determining step. Compared with nitrogen-doped carbon (NC) and tungsten carbide (W2C) decorated NC (W2C@NC), tungsten single atoms (WSA) and W2C decorated NC (WSA-W2C@NC) exhibited the lowest energy barrier for this step (0.943 eV), indicating more favorable reaction kinetics for long-chain to short-chain polysulfide conversion. Furthermore, using the CI-NEB method, they evaluated the kinetic barriers for K2S decomposition, which is a critical step in the charging process. The results showed that WSA-W2C@NC significantly reduces the decomposition energy barrier of K2S (0.32 eV) compared to W2C@NC (0.49 eV) and NC (0.97 eV), highlighting the catalytic role of WSA-W2C in promoting the decomposition of solid sulfides.
MD simulation
Theoretical foundations of MD
MD simulation is used for studying the short-timescale statistical properties and diffusion behavior of complex systems. Its foundation lies in Newtonian classical mechanics[47]. In a multi-particle system, each atom is regarded as a point mass whose motion follows Newton’s second law. The forces and accelerations acting on atoms can be described by potential gradients, atomic velocities, and atomic coordinates[47]:
where U is the energy of interatomic interactions,
Depending on how interatomic interactions are described, MD is divided into classical molecular dynamics (CMD) and ab initio molecular dynamics (AIMD)[69]. In CMD simulations, the potential energy governing atomic motion is known as potential energy functions or force fields[47]. For different simulation systems, the force field and its corresponding parameters are derived from empirical values or calculated using quantum mechanical methods. Commonly used force fields include AMBER[70], CHARMM[71], OPLS[72], GROMOS[73], and ReaxFF[74]. Specifically, AMBER was originally developed for simulating biomolecules such as proteins and nucleic acids, and it is now also used in electrolyte systems for K-S batteries[42]. CHARMM is designed for biomolecules and small organic molecules, and can be employed to model common electrolyte solvents[75]. GROMOS is mostly applied to biomolecules and gas-phase molecules, but studies have indicated that it does not perform well in simulating electrolyte systems[76]. The OPLS-AA version of the OPLS force field can reproduce experimental solubilities, thereby providing reliable simulations of ion diffusion behavior for K-S batteries[66]. The ReaxFF force field overcomes the limitation of traditional force fields that cannot simulate chemical reactions, which has been applied to simulate the lithiation process of sulfur cathodes in Li-S batteries[77], and is expected to find widespread applications in K-S batteries in the future. In contrast to CMD, AIMD simulations use methods such as the Hartree-Fock equation or DFT to solve the Schrödinger equation and obtain interatomic potential functions[57,78]. AIMD simulations can not only accurately handle all atoms in the periodic table but also model chemical reactions and polarization effects. Moreover, based on first-principles calculations, AIMD eliminates the drawbacks associated with empirical force fields. Nevertheless, inheriting the characteristics of first-principles calculations, while AIMD yields high-precision simulation results, the scale of simulatable systems and computational efficiency are significantly constrained.
Applications of MD for K-S batteries
MD simulation is an important method for probing the dynamic behavior of complex systems in K-S batteries. By capturing atomic positions and interactions over time, MD provides atomic-level insights into materials design and mechanistic understanding. The following sections illustrate different applications of MD simulations in K-S battery research, including ion transport properties in electrolytes, the dissolution behavior of polysulfide species, and solvation structure of electrolytes.
The dynamic behavior of ions and polysulfides in the bulk electrolyte affects the rate performance of K-S batteries[44]. MD simulations probe these dynamic processes by directly tracking the trajectories of atoms over time, enabling the quantification of transport properties including diffusion coefficients and ionic conductivity via mean square displacement (MSD) analysis[47]. Simultaneously, these simulations provide insights into the dissolution behavior of polysulfide intermediates, capturing their tendency to aggregate or disperse. For example, Wang et al.[66] systematically analyzed the dissolution and diffusion behavior of K2S in potassium acetate (KOAc) electrolytes of varying concentrations using MD simulations. Their simulations showed that, at a concentration of 30 m in KOAc electrolyte, the dissolution of K2S was significantly suppressed compared to dilute or pure water systems, primarily due to a substantial reduction in the diffusion coefficients of S2- and H2O. By calculating the MSD, they quantified the corresponding diffusion coefficients as 1.23 × 10-6 cm2 s-1 for S2- and 2.07 × 10-7 cm2 s-1 for H2O in the concentrated electrolyte, which is significantly lower than those in lower-concentration systems.
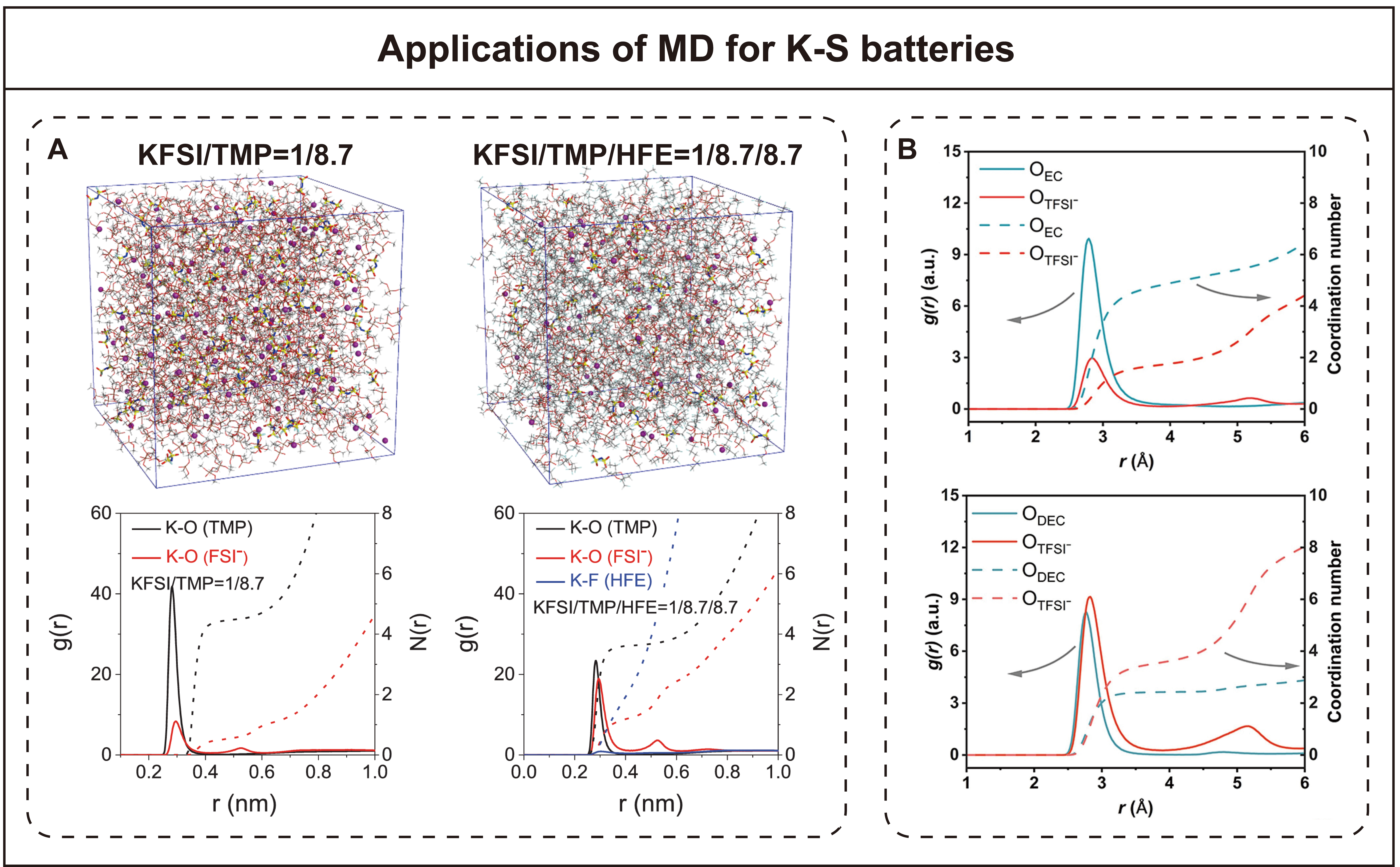
The solvation structure of K+ in electrolytes is essential for determining ion transport efficiency and interfacial stability[79]. Unlike DFT calculations, which mainly describe static electronic interactions, MD simulations capture the dynamic arrangement of solvent molecules and anions around K+. By analyzing radial distribution functions (RDFs) and coordination number distributions, MD reveals how electrolyte components affect the local environment of K+[80,81]. Li et al.[42] investigated the solvation structure of K+ in a nonflammable electrolyte composed of potassium bis(fluorosulfonyl)imide (KFSI) salt, trimethyl phosphate (TMP) flame-retardant solvent, and 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (HFE) diluent, as studied by MD simulation, as shown in Figure 6A. By analyzing RDFs and coordination number distributions, they demonstrated that the introduction of HFE weakens the K+-TMP interaction while enhancing the K+-FSI- interaction[42]. Specifically, the coordination number of TMP around K+ decreased from 4.47 to 3.64 upon HFE addition, whereas that of FSI- increased from 0.57 to 1.32, indicating a shift in the primary solvation sheath composition[42]. These changes were attributed to intermolecular interactions between TMP and HFE, which modulate the local coordination environment without direct K+-HFE coordination. Similarly, Yu and colleagues investigated the solvation structures of 3 M KTFSI in three carbonate solvents with different dielectric constants: EC, propylene carbonate (PC), and DEC [Figure 6B][82]. They revealed that EC, possessing the highest dielectric constant, successfully weakens the interaction between K+ and TFSI-, resulting in the highest solvent coordination number and the lowest anion coordination number among the three electrolytes. In contrast, DEC with the lowest dielectric constant exhibits the lowest solvent coordination number and the highest anion coordination number, indicating stronger ion pairing[82]. Based on the optimized solvation structure, the assembled K-S battery utilizing the
CONCLUSION AND OUTLOOK
In this review, we summarize the advances in K-S battery research, focusing on materials engineering and theoretical calculations. At the materials level, significant improvements have been achieved in the overall electrochemical performance of K-S batteries, with the core objective of mitigating the polysulfide shuttle. In addition to these experimental advances, theoretical simulations have provided crucial microscale insights. DFT calculations have provided atomic-scale insights into adsorption energy, reaction and diffusion barriers, and electronic structure characteristics, while MD simulations have elucidated dynamic processes including ion transport, polysulfide dissolution behavior, and solvation structure evolution.
Despite these significant advances, K-S batteries are still far from large-scale practical applications. The reported electrochemical performance remains unsatisfactory, and several deeply rooted challenges must be resolved. First, the severe shuttle effect caused by the high solubility of potassium polysulfides leads to continuous active material loss, rapid capacity decay, and corrosion of the potassium metal anode. Second, the highly reactive potassium metal anode suffers from an unstable SEI and pronounced dendrite growth, resulting in low Coulombic efficiency and serious safety hazards. Third, the large ionic radius of K+ induces substantial volume expansion during cycling, which compromises the structural integrity of host materials. These bottlenecks demand a concerted effort from both experimental materials innovation and theoretical simulation analysis. Several research directions that specifically target the above constraints deserve particular attention.
(1) Computational screening of catalytic hosts to suppress the shuttle effect
The severe polysulfide shuttle demands host materials that not only adsorb K2Sn effectively but also accelerate their catalytic conversion to short-chain products. Polar host materials with catalytic activity and moderate adsorption strength are promising candidates. Future work should combine high-throughput DFT calculations to evaluate adsorption energies and Gibbs free energy profiles of the sulfur reduction reaction for a large library of hosts. AIMD simulations can further elucidate the dynamic decomposition and conversion of polysulfides at the interface. Experimentally, these computational predictions should be validated by in situ Raman, X-ray diffraction and electrochemical testing, closing the loop between theory and experiment.
(2) Simulation-driven design of stable K metal anodes
The unstable SEI and dendrite growth on K metal can be addressed by optimizing electrolyte formulations and designing artificial SEI layers. MD simulations can analyze how different electrolyte compositions influence the microscopic solvation structure and the decomposition behavior at the electrode-electrolyte interface, thereby identifying stable SEI products and elucidating their passivation mechanisms. Through systematic MD screening of various component ratios and concentrations, optimal electrolyte formulations that form a dense and durable SEI can be identified. Meanwhile, DFT calculations can evaluate the potassiophilicity and the K+ diffusion barrier of candidate artificial SEI materials, enabling the selection of coatings that promote uniform K deposition.
(3) High-throughput computation and machine learning accelerated design of cathodes with high volume-expansion tolerance
The large ionic radius of K+ causes severe volume swings of electrodes. Materials solutions include hosts with tunable interlayer spacing (e.g., functionalized MXenes, expanded graphites, covalent-organic frameworks) and hierarchically porous architectures (e.g., metal-organic frameworks). High-throughput DFT calculations using the CI-NEB method can compute K+ diffusion barriers for hundreds of layered and porous materials, rapidly identifying fast ion conductors. Machine learning models based on geometric and electronic descriptors can then predict the volume expansion ratio and mechanical stability of candidate structures. Reactive MD simulations can further model the long-term structural evolution of the cathode under repeated expansion-contraction cycles, revealing failure mechanisms such as pore collapse or layer delamination.
DECLARATIONS
Authors’ contributions
Conceptualization, funding acquisition, and supervision: Liu, P.
Writing – original draft, review, and editing: Huang, M.
Data curation, literature analysis, and interpretation: Feng, X.; Shchurik, E. V.
Critical revision of the manuscript for important intellectual content: Xiang, H.; Shestakov, A. F.; Troshin, P.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20241754).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Braff, W. A.; Mueller, J. M.; Trancik, J. E. Value of storage technologies for wind and solar energy. Nat. Clim. Chang. 2016, 6, 964-9.
3. Zhu, Z.; Jiang, T.; Ali, M.; et al. Rechargeable batteries for grid scale energy storage. Chem. Rev. 2022, 122, 16610-751.
4. Ngoy, K. R.; Lukong, V. T.; Yoro, K. O.; et al. Lithium-ion batteries and the future of sustainable energy: a comprehensive review. Renew. Sustain. Energy. Rev. 2025, 223, 115971.
5. Jiang, T.; Shen, D.; Zhang, Z.; et al. Battery technologies for grid-scale energy storage. Nat. Rev. Clean. Technol. 2025, 1, 474-92.
6. Li, M.; Lu, J.; Chen, Z.; Amine, K. 30 years of lithium-ion batteries. Adv. Mater. 2018, 30, 1800561.
7. Khan, A.; Al Rashid, H.; Roy, P. K.; Chowdhury, S. I.; Sathi, S. A. Challenges and the way to improve lithium-ion battery technology for next-generation energy storage. Energy. Environ. Mater. 2025, 8, e70088.
8. Yao, W.; Liao, K.; Lai, T.; Sul, H.; Manthiram, A. Rechargeable metal-sulfur batteries: key materials to mechanisms. Chem. Rev. 2024, 124, 4935-5118.
9. Ma, L.; Lv, Y.; Wu, J.; Chen, Y.; Jin, Z. Recent advances in emerging non-lithium metal-sulfur batteries: a review. Adv. Energy. Mater. 2021, 11, 2100770.
10. Li, J.; Liang, G.; Zheng, W.; et al. Addressing cation mixing in layered structured cathodes for lithium-ion batteries: a critical review. Nano. Mater. Sci. 2023, 5, 404-20.
11. Prajapati A, Bhatnagar A. A review on anode materials for lithium/sodium-ion batteries. J. Energy. Chem. 2023, 83, 509-40.
12. Seh, Z. W.; Sun, Y.; Zhang, Q.; Cui, Y. Designing high-energy lithium-sulfur batteries. Chem. Soc. Rev. 2016, 45, 5605-34.
13. Shao, Y.; Pang, B.; Bird, L.; Robinson, J. B.; Shearing, P. R. Contemporary trends in lithium-sulfur battery design: a comparative review of liquid, quasi-solid, and all-solid-state architectures and mechanisms. Adv. Energy. Mater. 2025, 16, e03239.
14. Chen, Z.; Yu, Q.; Wang, W.; Zhang, J. Interface engineering strategies for shuttle mitigation in alkali metal-sulfur batteries: a comparative review from Li-S to Na-S and K-S systems. Nano. Micro. Lett. 2026, 18, 167.
15. Ding, J.; Zhang, H.; Fan, W.; Zhong, C.; Hu, W.; Mitlin, D. Review of emerging potassium-sulfur batteries. Adv. Mater. 2020, 32, 1908007.
16. Streng, R. L.; Steeger, T.; Senyshyn, A.; et al. A low-cost and high-energy aqueous potassium-ion battery. J. Energy. Chem. 2025, 106, 523-31.
17. Zhao, X.; Lu, Y.; Qian, Z.; Wang, R.; Guo, Z. Potassium-sulfur batteries: status and perspectives. EcoMat 2020, 2, e12038.
18. Liu, P.; Mitlin, D. Emerging potassium metal anodes: perspectives on control of the electrochemical interfaces. Acc. Chem. Res. 2020, 53, 1161-75.
19. Hosaka, T.; Kubota, K.; Hameed, A. S.; Komaba, S. Research development on K-ion batteries. Chem. Rev. 2020, 120, 6358-466.
20. Salado, M.; Amores, M.; Pozo-Gonzalo, C.; Forsyth, M.; Lanceros-Méndez, S. Advanced and sustainable functional materials for potassium-ion batteries. Energy. Mater. 2023, 3, 300037.
21. Lei, Y. J.; Yang, H. L.; Liang, Y.; et al. Progress and prospects of emerging potassium-sulfur batteries. Adv. Energy. Mater. 2022, 12, 2202523.
22. Wang, H.; Zhai, D.; Kang, F. Solid electrolyte interphase (SEI) in potassium ion batteries. Energy. Environ. Sci. 2020, 13, 4583-608.
23. Zhao, Q.; Hu, Y.; Zhang, K.; Chen, J. Potassium-sulfur batteries: a new member of room-temperature rechargeable metal-sulfur batteries. Inorg. Chem. 2014, 53, 9000-5.
24. Lu, X.; Bowden, M. E.; Sprenkle, V. L.; Liu, J. A low cost, high energy density, and long cycle life potassium-sulfur battery for grid-scale energy storage. Adv. Mater. 2015, 27, 5915-22.
25. Hwang, J.; Kim, H. M.; Yoon, C. S.; Sun, Y. Toward high-safety potassium-sulfur batteries using a potassium polysulfide catholyte and metal-free anode. ACS. Energy. Lett. 2018, 3, 540-1.
26. Xiong, P.; Han, X.; Zhao, X.; et al. Room-temperature potassium-sulfur batteries enabled by microporous carbon stabilized small-molecule sulfur cathodes. ACS. Nano. 2019, 13, 2536-43.
27. Ge, X.; Di, H.; Wang, P.; et al. Metal-organic framework-derived nitrogen-doped cobalt nanocluster inlaid porous carbon as high-efficiency catalyst for advanced potassium-sulfur batteries. ACS. Nano. 2020, 14, 16022-35.
28. Ye, C.; Shan, J.; Chao, D.; et al. Catalytic oxidation of K2S via atomic co and pyridinic N synergy in potassium-sulfur batteries. J. Am. Chem. Soc. 2021, 143, 16902-7.
29. Lee, S.; Park, H.; Rizell, J.; et al. High-energy and long-lifespan potassium-sulfur batteries enabled by concentrated electrolyte. Adv. Funct. Mater. 2022, 32, 2209145.
30. Ye, C.; Shan, J.; Li, H.; Kao, C. C.; Gu, Q.; Qiao, S. Z. Reducing overpotential of solid-state sulfide conversion in potassium-sulfur batteries. Angew. Chem. Int. Ed. 2023, 62, e202301681.
31. Song, W.; Yang, X.; Zhang, T.; et al. Optimizing potassium polysulfides for high performance potassium-sulfur batteries. Nat. Commun. 2024, 15, 1005.
32. Chen, X.; Yi, T.; Cen, Z.; et al. Organosulfur-rich porous carbon cathode enables soluble-polysulfide-free and high-rate potassium-sulfur batteries. Angew. Chem. Int. Ed. 2025, 65, e05751.
33. Yu, X.; Manthiram, A. A reversible nonaqueous room-temperature potassium-sulfur chemistry for electrochemical energy storage. Energy. Storage. Mater. 2018, 15, 368-73.
34. Liu, Y.; Wang, W.; Wang, J.; et al. Sulfur nanocomposite as a positive electrode material for rechargeable potassium-sulfur batteries. Chem. Commun. 2018, 54, 2288-91.
35. Hwang, J.; Kim, H. M.; Sun, Y. High performance potassium-sulfur batteries based on a sulfurized polyacrylonitrile cathode and polyacrylic acid binder. J. Mater. Chem. A. 2018, 6, 14587-93.
36. Ma, R.; Fan, L.; Wang, J.; Lu, B. Confined and covalent sulfur for stable room temperature potassium-sulfur battery. Electrochim. Acta. 2019, 293, 191-8.
37. Ma, S.; Zuo, P.; Zhang, H.; et al. Iodine-doped sulfurized polyacrylonitrile with enhanced electrochemical performance for room-temperature sodium/potassium sulfur batteries. Chem. Commun. 2019, 55, 5267-70.
38. Zhang, Y.; Lou, J.; Shuai, Y.; et al. A novel rechargeable potassium-sulfur battery based on liquid alloy anode. Mater. Lett. 2019, 242, 5-8.
39. Wang, L.; Bao, J.; Liu, Q.; Sun, C. Concentrated electrolytes unlock the full energy potential of potassium-sulfur battery chemistry. Energy. Storage. Mater. 2019, 18, 470-5.
40. Yao, Y.; Xu, R.; Chen, M.; et al. Encapsulation of SeS2 into nitrogen-doped free-standing carbon nanofiber film enabling long cycle life and high energy density K-SeS2 battery. ACS. Nano. 2019, 13, 4695-704.
41. Zhang, S.; Li, Z.; Li, M.; et al. Orbital electron coupling of Ga-Cd dual-atom sites catalyzes sulfur redox in potassium-sulfur battery. Nat. Commun. 2025, 16, 8652.
42. Li, Q.; Liu, G.; Kumar, P.; et al. Ultralow concentration nonflammable electrolytes mediated by intermolecular interactions for safer potassium-ion sulfur batteries. Adv. Funct. Mater. 2024, 35, 2416714.
43. Li, C.; Yang, D.; Yu, J.; et al. Three birds with one stone: multifunctional separators based on SnSe nanosheets enable high-performance Li-, Na- and K-sulfur batteries. Adv. Energy. Mater. 2024, 14, 2303551.
44. Chen, R.; Zhou, Y.; He, J.; Li, X. Advanced computational methods in lithium-sulfur batteries. Adv. Funct. Mater. 2024, 34, 2407986.
45. Huang, M.; Wang, Y.; Hou, Q. et al. Theoretical calculation and computational simulation on electrolyte for lithium metal battery. Prog. Chem. 2023, 35, 1847-63.
46. Feng, S.; Fu, Z. H.; Chen, X.; Zhang, Q. A review on theoretical models for lithium-sulfur battery cathodes. InfoMat 2022, 4, e12304.
47. Yao, N.; Chen, X.; Fu, Z.; Zhang, Q. Applying classical, ab initio, and machine-learning molecular dynamics simulations to the liquid electrolyte for rechargeable batteries. Chem. Rev. 2022, 122, 10970-1021.
48. Wang, X.; Fang, R.; Zhang, J.; et al. Dual-conductivity optimization toward high-rate and ultralong life all-solid-state lithium-sulfur batteries. Adv. Mater. 2026, 38, e22976.
49. Yuan, F.; Li, Z.; Zhang, D.; et al. Fundamental understanding and research progress on the interfacial behaviors for potassium-ion battery anode. Adv. Sci. 2022, 9, 2200683.
50. Hosaka, T.; Muratsubaki, S.; Kubota, K.; Onuma, H.; Komaba, S. Potassium metal as reliable reference electrodes of nonaqueous potassium cells. J. Phys. Chem. Lett. 2019, 10, 3296-300.
51. Xiao, N.; Mcculloch, W. D.; Wu, Y. Reversible dendrite-free potassium plating and stripping electrochemistry for potassium secondary batteries. J. Am. Chem. Soc. 2017, 139, 9475-8.
52. Shao, J.; Zheng, J.; Qin, L.; Zhang, S.; Ren, Y.; Wu, Y. K3SbS4 as a potassium superionic conductor with low activation energy for K-S batteries. Angew. Chem. Int. Ed. 2022, 61, e202200606.
54. Schrödinger, E. An undulatory theory of the mechanics of atoms and molecules. Phys. Rev. 1926, 28, 1049-70.
57. Kohn, W.; Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133-8.
58. Wang, L.; Zunger, A. Local-density-derived semiempirical pseudopotentials. Phys. Rev. B. 1995, 51, 17398-416.
59. Perdew, J. P.; Wang, Y. Pair-distribution function and its coupling-constant average for the spin-polarized electron gas. Phys. Rev. B. 1992, 46, 12947-54.
60. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
61. Perdew, J. P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B. 1992, 45, 13244-9.
62. Perdew, J. P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982-5.
63. Heyd, J.; Scuseria, G. E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207-15.
64. Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 2002, 98, 11623-7.
65. Liang, J.; Song, W.; Wang, H.; Ding, J.; Hu, W. Porosity engineering towards nitrogen-rich carbon host enables ultrahigh capacity sulfur cathode for room temperature potassium-sulfur batteries. Nanomaterials 2022, 12, 3968.
66. Wang, B.; Zhou, W.; Zhang, Y.; et al. An energetic K+-S aqueous battery with 96% sulfur redox utilization. Joule 2024, 8, 2033-48.
67. Zhang, Z. W.; Peng, H. J.; Zhao, M.; Huang, J. Q. Heterogeneous/Homogeneous mediators for high-energy-density lithium-sulfur batteries: progress and prospects. Adv. Funct. Mater. 2018, 28, 1707536.
68. Zhang, S.; Kong, Y.; Gu, Y.; et al. Strong d-π orbital coupling of Co-C4 atomic sites on graphdiyne boosts potassium-sulfur battery electrocatalysis. J. Am. Chem. Soc. 2024, 146, 4433-43.
69. Li, Y.; Zhao, Q.; Xing, Y.; et al. Multiscale simulation for electrolyte design: From microstructure-property decoding to macroscopic performance prediction. Energy. Storage. Mater. 2026, 86, 104927.
70. Wang, J.; Wolf, R. M.; Caldwell, J. W.; Kollman, P. A.; Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157-74.
71. Mackerell, A. D.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257-65.
72. Jorgensen, W. L.; Maxwell, D. S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225-36.
73. Hermans, J.; Berendsen, H. J. C.; Van, Gunsteren. W. F.; Postma, J. P. M. A consistent empirical potential for water-protein interactions. Biopolymers 2004, 23, 1513-8.
74. Chenoweth, K.; Van, Duin. A. C. T.; Goddard, W. A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A. 2008, 112, 1040-53.
75. Song, N.; Gao, Z.; Zhang, Y.; Li, X. B4C nanoskeleton enabled, flexible lithium-sulfur batteries. Nano. Energy. 2019, 58, 30-9.
76. García-Melgarejo, V.; Alejandre, J.; Núñez-Rojas, E. Parametrization with explicit water of solvents used in lithium-ion batteries: cyclic carbonates and linear ethers. J. Phys. Chem. B. 2020, 124, 4741-50.
77. Islam, M. M.; Ostadhossein, A.; Borodin, O.; et al. ReaxFF molecular dynamics simulations on lithiated sulfur cathode materials. Phys. Chem. Chem. Phys. 2015, 17, 3383-93.
78. Hartree, D. R. The wave mechanics of an atom with a non-coulomb central field. Part I. theory and methods. Math. Proc. Camb. Phil. Soc. 2008, 24, 89-110.
79. Xu, Y.; Ding, T.; Sun, D.; Ji, X.; Zhou, X. Recent advances in electrolytes for potassium-ion batteries. Adv. Funct. Mater. 2022, 33, 2211290.
80. Chen, J.; An, D.; Wang, S.; et al. Rechargeable potassium-ion full cells operating at -40 °C. Angew. Chem. Int. Ed. 2023, 62, e202307122.
81. Hu, Y.; Fu, H.; Geng, Y.; et al. Chloro-functionalized ether-based electrolyte for high-voltage and stable potassium-ion batteries. Angew. Chem. Int. Ed. 2024, 63, e202403269.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].