


MicroRNA-regulated mitochondrial dysfunction in heart failure
 0
0 Abstract
Heart failure (HF) is characterized by profound mitochondrial dysfunction, a central pathogenic mechanism driven by interconnected defects in metabolic dysregulation, excessive oxidative stress, impaired biogenesis, imbalanced dynamics, and defective mitophagy. This compromised mitochondrial fitness leads to bioenergetic deficiency, cardiomyocyte death, and progressive cardiac remodeling. MicroRNAs (miRNAs) have emerged as critical post-transcriptional regulators of mitochondrial homeostasis, capable of simultaneously modulating multiple components within these pathways. By fine-tuning the expression of key genes involved in fission/fusion, mitophagy, and biogenesis, miRNAs can either exacerbate or ameliorate HF progression, forming a complex and context-dependent regulatory network, and highlighting the potential of targeting the miRNA-mitochondria axis. However, clinical translation faces significant hurdles including target specificity, tissue-selective delivery and patient heterogeneity. Future research should focus on deciphering the mechanistic interplay between miRNA regulation and mitochondrial function in diverse HF contexts. This review aims to elucidate the molecular mechanisms by which miRNAs regulate mitochondrial dysfunction in HF, revealing novel therapeutic targets. A deeper understanding of this regulatory network is crucial for advancing HF management from symptom palliation toward mechanism-based precision medicine.
Keywords
INTRODUCTION
Heart Failure (HF) represents a clinical syndrome resulting from structural and/or functional impairments of the heart. It represents the common terminal stage of a wide spectrum of cardiovascular diseases, and is characterized clinically by symptoms such as dyspnea, fatigue, and systemic or pulmonary congestion[1]. Although recent advances in therapeutic interventions have significantly reduced acute mortality associated with many cardiovascular conditions, they have paradoxically contributed to a rising prevalence of chronic HF. Epidemiological data have shown that the prevalence of HF varies widely across populations, ranging between 1% and 3%, with a notably higher incidence among the elderly population aged 65 and above[2,3]. Given its profound impact on patients’ quality of life and its substantial global disease burden, HF has emerged as a major focus of research in cardiovascular medicine[4].
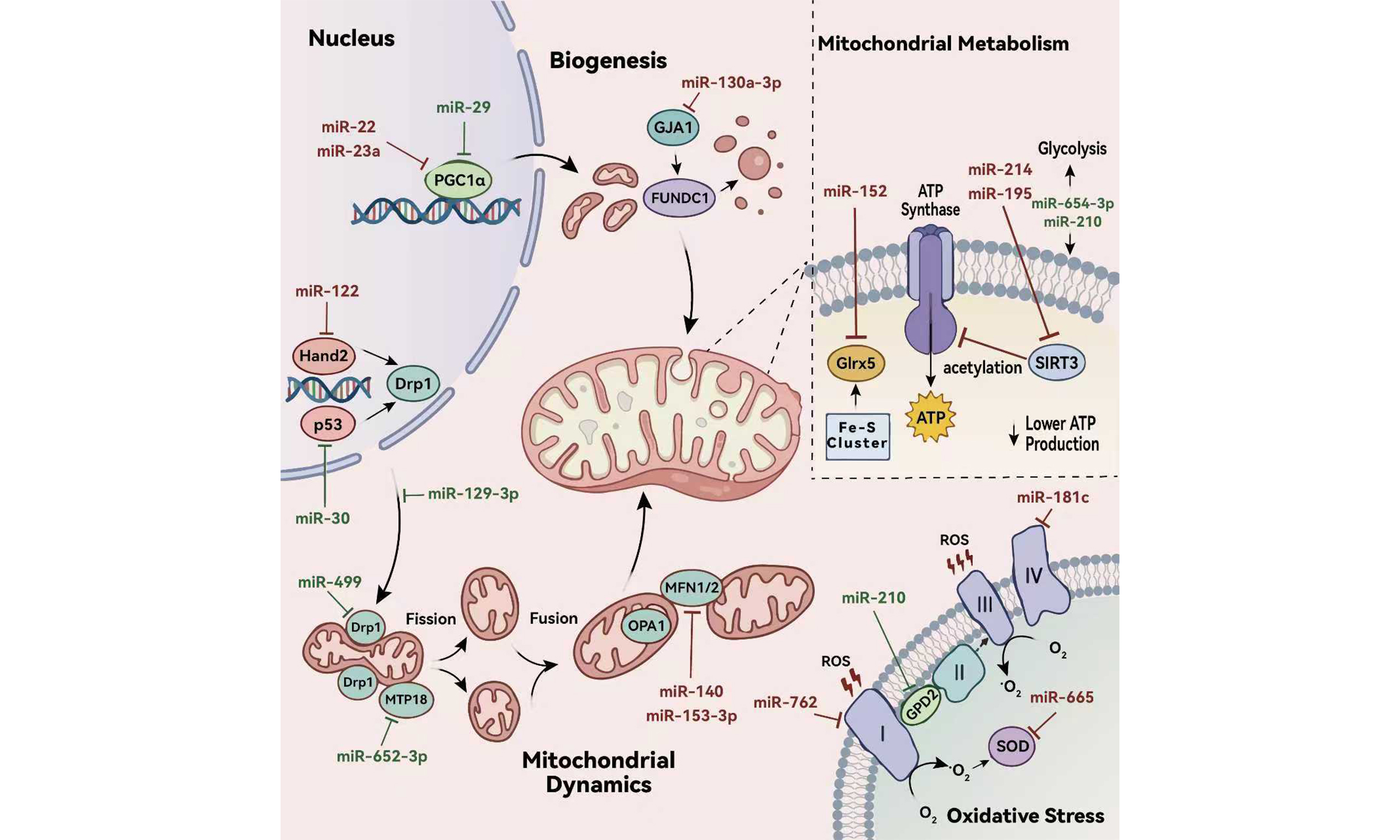
The pathogenesis of HF involves multiple interrelated mechanisms, including mitochondrial dysfunction, cardiac hypertrophy, endothelial dysfunction, myocardial fibrosis and inflammatory responses[5-9]. These pathways interact synergistically to drive the progression of HF. Among them, mitochondrial dysfunction plays a central role. It is characterized by metabolic derangements, excessive oxidative stress, impaired mitochondrial dynamics, and defective mitophagy[5]. Given the heart’s high energy demand, mitochondrial dysfunction critically undermines myocardial bioenergetics, leading to insufficient adenosine triphosphate (ATP) production. This energetic deficit directly compromises both systolic and diastolic function of cardiomyocytes, thereby initiating or exacerbating cardiac dysfunction in HF.
MicroRNAs (miRNAs) are a class of evolutionarily conserved, non-coding RNA (ncRNAs) molecules approximately 18~22 nucleotides in length. They modulate gene expression at the post-transcriptional level and are extensively implicated in a wide array of physiological processes and pathological conditions, including cardiovascular diseases[10-12]. Recent studies have demonstrated that under various pathological stimuli related to HF, including hypoxia/reperfusion (H/R) and hormonal changes, the expression of miRNAs becomes dysregulated. These miRNAs then act as regulatory nodes, critically influencing the pathogenesis of HF by targeting key pathways associated with mitochondrial dysfunction—such as mitochondrial biogenesis, dynamics (fusion and fission), and oxidative stress responses[13-15]. This review aims to provide a comprehensive overview of the molecular mechanisms through which miRNAs regulate mitochondrial dysfunction in the context of HF, highlighting their potential as both therapeutic targets and biomarkers[16,17].
MITOCHONDRIAL METABOLISMS
Metabolic abnormalities in HF
In the healthy myocardium, ATP is predominantly generated through mitochondrial oxidative phosphorylation (OXPHOS), which accounts for approximately 95% of total ATP production; the remaining ~ 5% is derived from glycolysis[18,19]. The reducing equivalents that fuel OXPHOS are primarily supplied by the oxidation of fatty acids, lactate, glucose, ketone bodies, and amino acids. Among these substrates, fatty acid oxidation contributes the largest share (40%~60%), followed by glucose oxidation (20%~40%)[20]. Critically, the healthy heart exhibits remarkable metabolic flexibility—dynamically shifting its substrate preference in response to changes in substrate availability, hormonal signals, and hemodynamic workload to meet its high energy demands[20].
In contrast, the failing heart undergoes profound alterations in energy metabolism, characterized by impaired metabolic flexibility, reduced overall ATP production, and a shift in substrate utilization. Although the precise nature of these metabolic changes remains somewhat controversial across different HF phenotypes, a consistent finding is the uncoupling of glycolysis from glucose oxidation—an inefficiency that compromises cardiac work efficiency regardless of whether glycolytic flux is increased or decreased.
Notably, Mericskay et al. reported divergent metabolic adaptations in HF with preserved ejection fraction (HFpEF) vs. HF with reduced ejection fraction (HFrEF)[21]. In HFpEF, they observed increased fatty acid oxidation coupled with reduced ketone body oxidation, contributing to diminished metabolic flexibility and cardiac efficiency. Conversely, in HFrEF, ketone body oxidation is upregulated—potentially as a compensatory mechanism to sustain energy production in the face of bioenergetic deficit[21].
miRNAs that exacerbate HF through inducing metabolic abnormalities
Zhang et al. reported elevated levels of miR-195 in the plasma, serum, and myocardial tissues of patients with advanced HF[22]. Subsequent work demonstrated that, in failing myocardium induced by transverse aortic constriction (TAC) or myocardial infarction (MI), miR-195 directly suppresses the expression of sirtuin 3 (SIRT3), a mitochondrial NAD+-dependent deacetylase. This downregulation leads to hyperacetylation of key enzymes involved in mitochondrial energy metabolism—most notably the pyruvate dehydrogenase complex (PDH) and ATP synthase. The resulting reduction in enzymatic activity impairs glucose oxidation, diminishes ATP production, and compromises mitochondrial respiratory capacity, thereby accelerating HF progression[22,23]. In a separate study,
Additionally, miR-152 is among the most significantly upregulated miRNAs in the human failing myocardium, and its expression is also markedly activated in animal models of cardiac pressure overload. Larocca et al. demonstrated that in HF mice induced by TAC, miR-152 disrupts myocardial iron homeostasis and impairs mitochondrial metabolism by directly targeting glutaredoxin 5 (Glrx5)—a critical regulator of iron-sulfur cluster biogenesis within mitochondria[24]. Notably, targeted inhibition of miR-152 in preclinical models attenuated adverse cardiac remodeling, slowed the decline in systolic function, and reduced ventricular dilation and myocardial fibrosis—highlighting its potential as a therapeutic target in HF[24].
miRNAs that attenuate HF through mitigating metabolic disturbances
Using integrated transcriptomic profiling and functional assays, Wu et al. demonstrated that miR-654-3p is significantly downregulated in MI tissue[25]. This reduction is associated with impaired mitochondrial energy metabolism and diminished ATP production. Importantly, restoration of miR-654-3p expression reversed these metabolic deficits and enhanced cardiac energy supply, underscoring its protective role in post-infarction remodeling[25]. Song et al. revealed that under ischemia/reperfusion (I/R) stress, miR-210 drives a metabolic shift in cardiomyocytes—from oxidative phosphorylation toward glycolysis[26]. This adaptive reprogramming appears to optimize cellular energy production and survival under hypoxic or stressful conditions, positioning miR-210 as a critical regulator of metabolic flexibility in the stressed heart[26].
The studies summarized above collectively demonstrate that miR-195, miR-214, and miR-152 are upregulated in HF and actively contribute to the deterioration of cardiac function by disrupting mitochondrial energy metabolism through distinct molecular mechanisms. In contrast, miR-654-3p and miR-210 appear to exert cardioprotective effects—either by restoring mitochondrial bioenergetics or by facilitating adaptive metabolic reprogramming under stress conditions. Although these miRNAs converge on the regulation of mitochondrial function, their individual targets and downstream pathways differ, underscoring the complexity and context-dependency of miRNA-mediated gene regulation in HF.
Moreover, the failing heart consistently exhibits dysregulation of multiple miRNAs, and experimental evidence shows that targeted modulation—either inhibition of detrimental miRNAs or restoration of beneficial ones—can ameliorate key features of HF across diverse preclinical models[16,27]. These findings highlight the dual potential of miRNAs as both sensitive biomarkers for disease stratification and promising therapeutic targets for precision intervention in HF.
MITOCHONDRIAL OXIDATIVE STRESS
Augmented oxidative stress in HF
Under physiological conditions, cardiomyocytes generate low levels of reactive oxygen species (ROS), which serve as signaling molecules in the regulation of various transcription factors and redox-sensitive pathways. These basal ROS levels are effectively neutralized by endogenous antioxidant systems, maintaining cellular redox homeostasis. In the failing heart, however, mitochondrial ROS production markedly exceeds the capacity of antioxidant defenses, resulting in oxidative stress. Elevated superoxide levels have been consistently documented across multiple experimental models of HF[28]. Excessive ROS accumulation not only damages lipids, proteins, and DNA but also disrupts calcium handling, thereby exacerbating cardiomyocyte injury. Mitochondrial electron transport chain complexes I, II, and III have been identified as the primary sites of pathological ROS generation in HF[29-31]. Among the key antioxidant enzymes, superoxide dismutase (SOD) plays a critical role in mitigating mitochondrial oxidative stress. Deficiency or inactivation of SOD has been linked to worsened mitochondrial dysfunction and accelerated progression of cardiac failure.
miRNAs that aggravate HF through aggravating mitochondrial oxidative stress
miR-181c, although transcribed in the nucleus, has been shown to localize to mitochondria, where it directly targets the mRNA of mitochondrial-encoded cytochrome c oxidase subunit 1 (mt-COX1). Das et al. discovered that overexpression of miR-181c in H9C2 cells causes structural remodeling and functional disruption of mitochondrial Complex IV by suppressing mt-COX1[32]. As the terminal part of Electron Transport Chain (ETC), Complex IV partial inhibition leads to electron congestion in upstream Complex, and directly reduces oxygen to superoxide, exacerbating oxidative stress[32]. Another study observed significant upregulation of miR-762 in myocardial tissue from I/R mice. MiR-762 directly inhibits the expression of NADH dehydrogenase subunit 2 (ND2), a core assembly subunit of Complex I. This inhibition disrupts electron flow through Complex I, leading to impaired ATP synthesis, increased ROS production, and further deterioration of cardiac function—highlighting miR-762 as another pathogenic miRNA that amplifies mitochondrial oxidative damage in HF[33].
Additionally, Lin et al. reported elevated expression of miR-665 in MI rats. MiR-665 was shown to directly suppress the glucagon-like peptide-1 receptor (GLP1R), leading to inactivation of the cAMP signaling pathway and abnormal expression of downstream proteins, especially SOD[34]. The consequent reduction in SOD activity exacerbates oxidative stress, contributing to myocardial injury and dysfunction. Importantly, administration of a miR-665 inhibitor in the same HF model effectively restored GLP1R signaling, normalized SOD levels, and ameliorated cardiac remodeling and functional decline—highlighting miR-665 as a potential therapeutic target in HF[34].
miRNAs that alleviate HF through alleviating mitochondrial oxidative stress
Song et al. demonstrated that miR-210 exerts cardioprotective effects, in part, by suppressing mitochondrial reactive oxygen species (mtROS) production in I/R myocardium[26]. A key target of miR-210 is glycerol-3-phosphate dehydrogenase 2 (GPD2), a mitochondrial enzyme that plays a dual role in the glycerol phosphate shuttle and as a component of the electron transport chain. GPD2 has been identified as a significant source of mtROS under pathological conditions. By downregulating GPD2 expression, miR-210 reduces electron leakage from the respiratory chain, thereby attenuating oxidative stress and preserving mitochondrial integrity and function[26].
Notably, beyond its well-established role in regulating mitochondrial energy metabolism, miR-210 also demonstrates a potent ability to modulate oxidative stress—highlighting its multifaceted influence on cardiac homeostasis. This dual functionality suggests that miR-210 may occupy an upstream regulatory node within the molecular network governing HF pathogenesis, enabling it to coordinately influence both bioenergetic and redox pathways. Given its capacity to enhance metabolic adaptability while simultaneously mitigating oxidative damage, we propose that miR-210 represents a particularly promising and high-priority candidate for the development of miRNA-based therapeutics in HF.
MITOCHONDRIAL BIOGENESIS
Impaired mitochondrial biogenesis in HF
Mitochondrial biogenesis—the process by which cells generate new mitochondria to maintain organelle function and meet energy demands—is essential for sustaining cardiac bioenergetics. Impairment of this process compromises ATP production and disrupts the energetic homeostasis. A central regulator that upregulates mitochondrial biogenesis is peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α). And the activity of PGC-1α is tightly modulated by post-translational modifications: it is activated via phosphorylation by AMP-activated protein kinase (AMPK) and deacetylation by sirtuin 1 (SIRT1)[35]. Notably, studies by Whittington et al. and Li et al. indicated that both the expression and activity of PGC-1α are reduced in HF[36,37]. Importantly, interventions that directly or indirectly enhance PGC-1α levels have consistently shown therapeutic benefits across diverse preclinical HF models. These findings firmly establish PGC-1α as a master regulator of mitochondrial biogenesis and a pivotal node in both the pathogenesis of HF and the development of potential metabolic therapies.
miRNAs that exacerbate HF via impairing mitochondrial biogenesis
Study has demonstrated that miR-22 directly targets the 3’ untranslated region (3’UTR) of PGC-1α mRNA, leading to its post-transcriptional repression and reduced protein expression in I/R rats[38]. Sun et al. observed myocardial injury in ovariectomy-induced estrogen-deficient mice and clarified that this outcome was partially due to miR-23a targeting PGC-1α via the same mechanism[39]. In addition, Du et al. reported that miR-22 also suppresses SIRT1 expression, thereby further inhibiting PGC-1α activity through the SIRT1-PGC-1α signaling axis[38]. Given that PGC-1α is a master regulator of mitochondrial biogenesis, the downregulation of this pathway by miR-22 and miR-23a results in impaired mitochondrial renewal, diminished oxidative capacity, and compromised bioenergetic support in cardiomyocytes—ultimately accelerating myocardial injury.
miRNAs that mitigate HF via promoting mitochondrial biogenesis
Caravia et al. reported that, under physiological conditions, miR-29 fine-tunes mitochondrial biogenesis by moderately repressing PGC-1α expression and plays a protective role[40]. In this context, loss of miR-29 leads to unchecked PGC-1α overexpression, which may drive aberrant mitochondrial biogenesis. Paradoxically, this results in the accumulation of structurally and functionally impaired mitochondria, disrupts cellular energy balance, and ultimately exacerbates HFpEF[40].
This apparent contradiction—where both miR-22/23a and miR-29 target PGC-1α yet exert opposing effects on HF progression—has drawn our particular attention. We hypothesize that miR-29, while suppressing biogenesis, simultaneously promotes mitochondrial quality control (e.g., through enhanced mitophagy or improved proteostasis), yielding a net protective effect. In contrast, miR-22 and miR-23a appear to suppress biogenesis without concomitant enhancement of quality mechanisms, leading to energetic insufficiency and cardiomyocyte dysfunction. These observations underscore that the functional outcome of modulating mitochondrial biogenesis depends not only on the quantity of mitochondria generated but also on their functional integrity. However, direct experimental validation for this hypothesis is currently lacking. Future research should address the unknown influence of miRNAs on mitochondrial quality to provide a more comprehensive understanding of the opposing effects on HF progression.
Moreover, while current research has predominantly centered on the PGC-1α axis as the primary regulator of biogenesis, the broader regulatory landscape likely involves additional miRNA-mediated pathways that remain underexplored. Future studies should aim to delineate these alternative mechanisms to fully elucidate how miRNAs orchestrate the delicate balance between mitochondrial quantity, quality, and function in the failing heart.
MITOCHONDRIAL DYNAMICS
Modifications in mitochondrial dynamics in HF
Mitochondria are highly dynamic organelles that continuously undergo fusion and fission to maintain their morphology, subcellular distribution, functional integrity, and homeostatic abundance. Mitochondrial fission is primarily regulated by dynamin-related protein 1 (Drp1). Upon activation, Drp1 translocates to the outer mitochondrial membrane (OMM), where it is recruited by adaptor proteins, and then initiates fission[41]. During mitochondrial fusion, OMM fusion occurs first, mediated by proteins such as Mitofusin 1/2 (Mfn1/2), and followed by inner mitochondrial membrane (IMM) fusion, which is mainly mediated by Optic atrophy 1 (Opa1)[41,42]. Additionally, Opa1 also plays a critical role in maintaining cristae architecture and facilitating the proper assembly and stability of respiratory chain supercomplexes. Dysregulated mitochondrial dynamics are a hallmark of the failing heart. In HF, levels of fusion-related proteins often decrease while fission-related protein levels typically increase, collectively leading to excessive mitochondrial fission and a fragmented morphology[43,44].
miRNAs that worsen HF via inducing abnormalities in dynamics
As previously discussed, mitochondrial dynamics are profoundly disrupted in HF, typically manifesting as excessive fission and impaired fusion. Studies have confirmed that under I/R- or drug-induced cardiac dysfunction, expression levels of miR-140, miR-153-3p, and miR-122 in cardiomyocytes are significantly elevated[45-47]. Among these, miR-140 and miR-153-3p contribute to mitochondrial dysfunction in HF by targeting Mfn1, while miR-122 upregulates Drp1 expression by directly inhibiting the transcription factor Hand2. And this leads to excessive mitochondrial fission, induces cardiomyocyte apoptosis, and ultimately amplifies the progression of the HF phenotype[45-47].
miRNAs that attenuate HF via maintaining mitochondrial dynamics homeostasis
Several studies have demonstrated that miR-30, miR-129-3p, and miR-499 exert cardioprotective effects respectively in HF models induced by H2O2, Ang-(1-9), and I/R by converging on Drp1, yet each operates through distinct molecular mechanisms. Li et al. reported that miR-30 attenuates Drp1 expression indirectly by targeting tumor protein p53 mRNA, thereby suppressing the p53-Drp1 signaling axis[48]. In contrast, miR-129-3p targets protein kinase A inhibitor (PKIA), thereby lifting the inhibitory effect of PKIA on PKA. This activation of PKA signaling enhances phosphorylation of Drp1 at Ser637, which inhibits its function[49]. Another study revealed that miR-499 directly targets the catalytic subunit of calcineurin to reduce its level and activity. This results in decreased dephosphorylation of Drp1 at Ser656, consequently suppressing Drp1 translocation from the cytoplasm to the OMM[50].
In addition to miRNAs that modulate Drp1, Wang et al. further identified miR-652-3p as a regulator of mitochondrial fission[51]. This miRNA effectively suppresses mitochondrial division by directly targeting and inhibiting mitochondrial protein 18 kDa (MTP18), a protein that promotes mitochondrial fission[51].
Current research on miRNA-mediated regulation of mitochondrial dynamics has primarily focused on a limited set of targets, notably Drp1 and Mfn1. However, the processes of mitochondrial fusion and fission are highly complex, involving coordinated actions of numerous proteins. Whether miRNAs can target other components within these pathways to fine-tune mitochondrial dynamics remains an open question worthy of further investigation.
Moreover, alterations in mitochondrial dynamics influence not only organelle quantity but also qualitative aspects. Therefore, functional assessments of mitochondria following fusion or fission events are essential. Future studies should determine whether newly formed mitochondria can properly execute their physiological functions, particularly in energy production and cellular homeostasis, to fully explain the functional consequences of miRNA-mediated regulation.
MITOPHAGY
Impaired mitophagy in HF
Mitophagy represents a selective autophagic process essential for removing damaged mitochondria, thereby preserving mitochondrial fitness. As a critical component of mitochondrial quality control, appropriate mitophagy exerts a protective effect on cellular homeostasis. In cardiomyocytes, mitophagy is primarily mediated by the ubiquitin-dependent PINK1/Parkin pathway, which is considered the canonical mechanism[52]. Additionally, receptor proteins like PTEN-induced putative kinase 1 (FUNDC1), BCL2-interacting protein 3 (BNIP3), and Nip3-like protein X (NIX) activate alternative, ubiquitin-independent pathways[53]. In the context of HF, clinical evidence indicates impaired mitophagy. For instance, Billia et al. found decreased PINK1 protein expression and reduced mitophagy levels in advanced HF patients, whereas pharmacological augmentation of autophagy improved cardiac function[54]. Similarly, myocardial tissue from HF patients exhibits significantly downregulated FUNDC1 expression. Experimental upregulation of the FUNDC1-mediated mitophagy has been proven protective for the failing heart[55]. Collectively, these studies underscore that maintaining mitophagy balance is crucial for ensuring mitochondrial homeostasis, highlighting its potential as a therapeutic target for HF.
miRNAs that promote HF via reducing mitophagy
Gap junction protein alpha 1 (GJA1) is recognized as a key regulator of mitochondrial function, in part through its ability to activate FUNDC1-mediated mitophagy. Yan et al. discovered significantly elevated expression of miR-130a-3p in both myocardial tissue from I/R rats and blood from acute myocardial infarction (AMI) patients[56]. Mechanistically, miR-130a-3p directly targets and suppresses GJA1 expression. This inhibition thereby obstructs mitophagy, preventing the effective clearance of damaged mitochondria, and ultimately exacerbating myocardial injury[56].
miRNAs that mitigate HF via controlling mitophagy
Mitochondrial homeostasis depends on a balance between clearance and biogenesis, which could be disrupted by uncontrolled mitophagy. This results in a state of mitochondrial suboptimal state, potentially accelerating the progression of diseases[57,58]. Mu et al. revealed the loss of miR-494-3p during H/R injury relieves its inhibitory effect on the target gene PGC-1α. The subsequent upregulation of PGC-1α activates the PINK/Parkin-mediated mitophagy, ultimately bringing about excessive mitophagy[59]. Importantly, exogenous administration of miR-494-3p can reverse this process, inhibit excessive mitophagy, and exert a cardioprotective effect[59]. Current understanding of how specific miRNAs govern mitophagy in HF remains incomplete, highlighting a critical gap in knowledge that warrants focused investigation. Furthermore, mitophagy represents a highly dynamic process. Pathological outcomes can arise not only from its impairment but equally from its overactivation, each contributing to mitochondrial insufficiency. This highlights the importance of developing strategies to modulate mitophagy within an optimal range, aiming to restore physiological balance rather than blanket inhibition or promotion.
TRANSLATIONAL CHALLENGES AND FUTURE DIRECTIONS
Substantial preclinical studies in HF models have demonstrated that therapeutic strategies utilizing miRNA mimics, vector-mediated overexpression of protective miRNAs or silencing of upregulated miRNAs through antisense oligonucleotides (e.g., antagomirs) or locked nucleic acids (LNA), can effectively alleviate mitochondrial dysfunction and subsequently reverse cardiac impairment[60]. Consequently, miRNA-based therapies targeting mitochondrial dysfunction are garnering significant attention. Despite this compelling promise, no miRNA-targeted therapy has yet advanced to clinical approval for HF treatment, highlighting a substantial translational gap between basic research and clinical application.
A primary obstacle is to find a safe, efficient, and cardiac-selective delivery of miRNA therapeutics. So far, lipid nanoparticles (LNPs) represent one of the most advanced delivery platforms. The Food and Drug Administration (FDA) has approved siRNA drugs based on LNP delivery system, demonstrating the clinical feasibility of this approach for ncRNAs therapeutics[61]. Similarly, adeno-associated viruses (AAVs), valued for their low immunogenicity and capacity for sustained transgene expression, have emerged as a major vector for cardiovascular gene therapy. Several AAV-based gene therapies for HF have entered clinical trials, providing valuable technical experience for future miRNA delivery[61]. Beyond these established systems, novel delivery platforms continue to emerge, including polymeric nanoparticles, exosomes, and aptamer- or peptide-based targeted delivery systems. These advancements in delivery technology are laying a critical foundation for translating miRNA therapies from concept to clinical reality.
Another significant challenge in miRNA therapy involves off-target effects and overall safety concerns. Given that a single miRNA can regulate multiple target genes, its pleiotropic nature raises the potential for unintended downstream consequences, which could lead to adverse effects. Therefore, enhancing cardiac tropism becomes a primary focus. One potential approach is to engineer vectors to evade non-target tissues. For instance, incorporating liver-specific miR-122 target sequences into the vector’s 3’UTR can selectively suppress off-target expression in liver[62]. Alternatively, LNP with the absence of plasma apolipoprotein E enables to circumvent the dominant hepatic clearance pathway and allows the inherent cardiotropic property of LNP to be unmasked[63]. Furthermore, the use of cardiac-specific promoters, such as the alpha myosin heavy chain (αMHC or Myh6) and the cardiac troponin T promoter (cTnT or TnnT2),can restrict expression to cardiomyocytes[64]. Alongside targeted delivery, the inherent polygenic nature of miRNA regulation necessitates rigorous safety profiling. To mitigate this safety risk, comprehensive safety assessments in large animal models are essential prior to clinical translation, ensuring a thorough evaluation of potential toxicities.
The inherent heterogeneity of HF presents an additional layer of complexity, as miRNA expression profiles vary across etiologies, stages, and phenotypic subtypes. This variability precludes a universal therapeutic strategy and necessitates a paradigm shift from a one-size-fits-all approach to biomarker-guided precision medicine. Therefore, to effectively target the miRNA-mitochondria axis, future efforts must integrate multi-omics profiling (e.g., tissue and plasma miRNA signature, metabolomic and proteomic data) with deep clinical phenotyping (e.g., imaging, hemodynamics, comorbidities) to identify distinct patient endotypes. This stratification will enable the matching of specific miRNA-targeted therapies to the responsive patients most likely to benefit. For instance, therapies targeting miR-130a-3p or miR-494-3p might be prioritized for HF patients with imbalance of mitophagy, whereas interventions modulating miR-210 could be tailored to those with a specific metabolic signature. The development and validation of such companion diagnostics are indispensable for designing successful clinical trials and realizing the promise of precise miRNA therapeutics in HF.
However, translating miRNA signatures into reliable clinical biomarkers faces several practical challenges. First, the level of circulating miRNAs can be influenced by pre-analytical variables such as sample collection tubes, processing time, and hemolysis, necessitating strict protocol standardization. Second, miRNA expression exhibits dynamic changes across HF stages, complicating the interpretation of a single measurement. Finally, the lack of harmonization in detection platforms (e.g., qPCR, RNA-seq, microarray) and data normalization methods across studies hinders large-scale clinical adoption. Overcoming these issues is crucial for developing robust miRNA-based companion diagnostics.
CONCLUSION
HF is a complex and heterogeneous clinical syndrome whose pathogenesis is intimately linked to mitochondrial dysfunction. As discussed, this dysfunction in HF arises from interconnected alternations in substrate metabolism, excessive oxidative stress, insufficient biogenesis, imbalanced dynamics, and defective mitophagy. Critically, miRNAs have emerged as pivotal post-transcriptional regulators of these processes, fine-tuning the expression of key genes governing mitochondrial structure, function, and quality control. Collectively, these regulations form a complex regulatory network characterized by both the cooperation of multiple miRNAs on shared targets and the pleiotropic regulation by individual miRNAs (e.g., miR-210). This precise and holistic system coordinately governs mitochondrial function and contributes to HF progression [Table 1].
miRNAs that promote and attenuate HF with their targets and potential mechanisms
| MicroRNAs | Targets | Mechanisms | Experimental models | References |
| Exacerbate HF | ||||
| miR-140 | Mfn1 | Fission | I/R mice | [47] |
| miR-22 | SIRT1 PGC-1α | Biogenesis | I/R rats | [39] |
| miR-122 | Hand2 | Fission | miR-122-induced HF mice | [46] |
| miR-152 | Glrx5 | Fe-S cluster | human biopsy TAC mice | [24] |
| miR-214 | SIRT3 | glucose oxidation | Ang II-treated mice | [23] |
| miR-665 | GLP1R | cAMP/SOD | MI rats | [34] |
| miR-130a-3p | GJA1 | Mitophagy | human biopsy I/R rats | [56] |
| miR-153-3p | Mfn1 | Fission | ISO-treated mice | [45] |
| miR-181c | mt-COX1 | ETC | miR-181c-induced HF rats | [32] |
| miR-195 | SIRT3 | glucose oxidation | human biopsy TAC mice MI mice | [22] |
| miR-762 | ND2 | ETC | I/R mice | [33] |
| miR-23a | PGC-1α | Biogenesis | OVX mice | [38] |
| Attenuate HF | ||||
| miR-29 | PGC-1α | Biogenesis | miR-29 KO mice | [40] |
| miR-652-3p | MTP18 | Fission | I/R mice | [51] |
| miR-30 | p53 | Fission | H2O2-treated NRCMs | [48] |
| miR-129-3p | PKIA-Drp1 | Fission | Ang-(1-9)-treated NRCMs | [49] |
| miR-210 | GPD2 - | ETC glycolysis | I/R mice | [26] |
| miR-499 | Calcineurin | fission | I/R mice | [50] |
| miR-654-3p | - | glycolysis | MI mice | [25] |
| miR-494-3p | PGC-1α | Mitophagy | H/R H9C2 cells | [59] |
Consequently, targeting the miRNA-mitochondria axis represents a promising frontier for HF therapies, with the potential to move beyond symptomatic management toward mechanism-based, precision medicine. However, significant challenges remain—including achieving cardiac-specific delivery, minimizing off-target effects, and addressing the molecular heterogeneity across HF subtypes. Future efforts must focus on elucidating context-specific miRNA-mitochondria interactions, leveraging advanced multi-omics profiling for patient stratification strategies, and developing targeted delivery platforms. Success in this endeavor could facilitate a paradigm shift in HF treatment, moving from palliative support to disease modification in HF.
DECLARATIONS
Authors’ contributions
Writing - original draft: Wang Y, Yang F
Visualization: Yang F, Cui J
Data curation, investigation: Zuo Y, Zheng Y, Luo Z, Wang Y, Yang F
Formal analysis: Liu Q, Chen R
Conceptualization, supervision, writing - review & editing: Xu X, Xia Y
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool DeepSeek (version 3.2, released 2025-12-01) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the Program for National Science Funds of China (Grant No. 82370386) and Special Initiative for Advancing Innovative Medical Research Xijing Hospital (XJZT25CX43).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Bozkurt B, Coats AJ, Tsutsui H, et al. Universal definition and classification of heart failure: a report of the heart failure society of america, heart failure association of the european society of cardiology, japanese heart failure society and writing committee of the universal definition of heart failure. J Card Fail. 2021;27:387-413.
2. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res. 2022;118:3272-87.
3. Emmons-Bell S, Johnson C, Roth G. Prevalence, incidence and survival of heart failure: a systematic review. Heart. 2022;108:1351-60.
4. Lin Q, Lv Z, Li D, et al. Heart failure medication treatment and prognosis: a retrospective cross-sectional study. Front. Pharmacol. 2025;16:1532123.
5. Brown DA, Perry JB, Allen ME, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2016;14:238-50.
7. Alcaide P, Kallikourdis M, Emig R, Prabhu SD. Myocardial inflammation in heart failure with reduced and preserved ejection fraction. Circ Res. 2024;134:1752-66.
8. Tsigkou V, Oikonomou E, Anastasiou A, et al. Molecular mechanisms and therapeutic implications of endothelial dysfunction in patients with heart failure. Int J Mol Sci. 2023;24:4321.
9. González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. 2018;71:1696-706.
10. Vishnoi A, Rani S. MiRNA biogenesis and regulation of diseases: an overview. Methods Mol Biol. 2017;1509:1-10.
12. Alaei A, Kakumani PK. MicroRNA chemical modifications in post-transcriptional gene silencing and human diseases. Mol Ther Nucleic Acids. 2025;36:102745.
13. Ao X, Ding W, Li X, et al. Non-coding RNAs regulating mitochondrial function in cardiovascular diseases. J Mol Med. 2023;101:501-26.
14. Zhang G, Wang S, Chen Y, et al. MicroRNAs regulating mitochondrial function in cardiac diseases. Front Pharmacol. 2021;12:663322.
15. Klimczak-Tomaniak D, Haponiuk-Skwarlińska J, Kuch M, Pączek L. Crosstalk between microRNA and oxidative stress in heart failure: a systematic review. Int J Mol Sci. 2022;23:15013.
16. Qian L, Zhao Q, Yu P, et al. Diagnostic potential of a circulating miRNA model associated with therapeutic effect in heart failure. J Transl Med. 2022;20:267.
17. Maciejak A, Kostarska-srokosz E, Gierlak W, et al. Circulating miR-30a-5p as a prognostic biomarker of left ventricular dysfunction after acute myocardial infarction. Sci Rep. 2018;8:9883.
18. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. 2021;128:1487-513.
19. Pham T, Loiselle D, Power A, Hickey AJR. Mitochondrial inefficiencies and anoxic ATP hydrolysis capacities in diabetic rat heart. Am J Physiol Cell Physiol. 2014;307:C499-507.
20. Sun Q, Karwi QG, Wong N, Lopaschuk GD. Advances in myocardial energy metabolism: metabolic remodelling in heart failure and beyond. Cardiovasc Res. 2024;120:1996-2016.
21. Mericskay M, Zuurbier CJ, Heather LC, et al. Cardiac intermediary metabolism in heart failure: substrate use, signalling roles and therapeutic targets. Nat Rev Cardiol. 2025;22:704-27.
22. Zhang X, Ji R, Liao X, et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. 2018;137:2052-67.
23. Ding Y, Zhang Y, Lu J, et al. MicroRNA-214 contributes to Ang II-induced cardiac hypertrophy by targeting SIRT3 to provoke mitochondrial malfunction. Acta Pharmacol Sin. 2020;42:1422-36.
24. Larocca TJ, Seeger T, Prado M, et al. Pharmacological silencing of MicroRNA-152 prevents pressure overload-induced heart failure. Circ Heart Failure. 2020;13:e006298.
25. Wu C, Zhang X, Chen L, et al. Pyroptosis and mitochondrial function participated in miR-654-3p-protected against myocardial infarction. Cell Death Dis. 2024;15:393.
26. Song R, Dasgupta C, Mulder C, Zhang L. MicroRNA-210 controls mitochondrial metabolism and protects heart function in myocardial infarction. Circulation. 2022;145:1140-53.
27. Ding H, Wang Y, Hu L, et al. Combined detection of miR-21-5p, miR-30a-3p, miR-30a-5p, miR-155-5p, miR-216a and miR-217 for screening of early heart failure diseases. Biosci Rep. 2020;40:BSR20191653.
28. Dey S, Demazumder D, Sidor A, Foster DB, O’Rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. 2018;123:356-71.
29. Kuka S, Tatarkova Z, Racay P, Lehotsky J, Dobrota D, Kaplan P. Effect of aging on formation of reactive oxygen species by mitochondria of rat heart. Gen Physiol Biophys. 2014;32:415-20.
30. Korge P, John SA, Calmettes G, Weiss JN. Reactive oxygen species production induced by pore opening in cardiac mitochondria: the role of complex II. J Biol Chem. 2017;292:9896-905.
31. Madungwe NB, Zilberstein NF, Feng Y, Bopassa JC. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. Am J Cardiovasc Dis. 2016;6:93-108.
32. Das S, Bedja D, Campbell N, et al. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS ONE. 2014;9:e96820.
33. Yan K, An T, Zhai M, et al. Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. 2019;10:500.
34. Lin B, Feng DG, Xu J. microRNA‐665 silencing improves cardiac function in rats with heart failure through activation of the cAMP signaling pathway. J Cell Physiol. 2019;234:13169-81.
35. Qian L, Zhu Y, Deng C, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Sig Transduct Target Ther. 2024;9:50.
36. Li Y, Feng Y, Liu X, et al. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 2021;38:101771.
37. Whittington HJ, Harding I, Stephenson CI, et al. Cardioprotection in the aging, diabetic heart: the loss of protective Akt signalling. Cardiovasc Res. 2013;99:694-704.
38. Du JK, Cong BH, Yu Q, et al. Upregulation of microRNA-22 contributes to myocardial ischemia-reperfusion injury by interfering with the mitochondrial function. Free Radical Biol Med. 2016;96:406-17.
39. Sun L, Wang N, Ban T, et al. MicroRNA-23a mediates mitochondrial compromise in estrogen deficiency-induced concentric remodeling via targeting PGC-1α. J Mol Cell Cardiol. 2014;75:1-11.
40. Caravia XM, Fanjul V, Oliver E, et al. The microRNA-29/PGC1α regulatory axis is critical for metabolic control of cardiac function. PLoS Biol. 2018;16:e2006247.
41. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol Mech Dis. 2020;15:235-59.
42. Anand R, Wai T, Baker MJ, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919-29.
43. Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91-9.
44. Hu L, Ding M, Tang D, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. 2019;9:3687-706.
45. Wang T, Zhai M, Xu S, et al. NFATc3-dependent expression of miR-153-3p promotes mitochondrial fragmentation in cardiac hypertrophy by impairing mitofusin-1 expression. Theranostics. 2020;10:553-66.
46. Shi Y, Zhang Z, Yin Q, et al. Cardiac‐specific overexpression of miR‐122 induces mitochondria‐dependent cardiomyocyte apoptosis and promotes heart failure by inhibiting Hand2. J Cell Mol Med. 2021;25:5326-34.
47. Li J, Li Y, Jiao J, et al. Mitofusin 1 is negatively regulated by MicroRNA 140 in cardiomyocyte apoptosis. Mol Cell Biol. 2023;34:1788-99.
48. Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6:e1000795.
49. Sotomayor-Flores C, Rivera-Mejías P, Vásquez-Trincado C, et al. Angiotensin-(1-9) prevents cardiomyocyte hypertrophy by controlling mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death Differ. 2020;27:2586-604.
50. Wang J, Jiao J, Li Q, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. 2010;17:71-8.
51. Wang K, Gan T, Li N, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017;24:1111-20.
52. Lin J, Chen K, Chen W, et al. Paradoxical mitophagy regulation by PINK1 and TUFm. Mol Cell. 2020;80:607-20.e12.
53. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24:787-95.
54. Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. 2011;108:9572-7.
55. Wu S, Lu Q, Wang Q, et al. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017;136:2248-66.
56. Yan Y, Tian L, Jia Q, et al. MiR-130a-3p regulates FUNDC1-mediated mitophagy by targeting GJA1 in myocardial ischemia/reperfusion injury. Cell Death Discov. 2023;9:77.
57. Gustafsson ÅB, Dorn GW. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol Rev. 2019;99:853-92.
58. Wang S, Long H, Hou L, et al. The mitophagy pathway and its implications in human diseases. Sig Transduct Target Ther. 2023;8:304.
59. Mu N, Zhang T, Zhu Y, Lu B, Zheng Q, Duan J. The mechanism by which miR-494-3p regulates PGC1-α-mediated inhibition of mitophagy in cardiomyocytes and alleviation of myocardial ischemia—reperfusion injury. BMC Cardiovasc Disord. 2023;23:204.
60. Mckinsey TA, Foo R, Anene-Nzelu CG, et al. Emerging epigenetic therapies of cardiac fibrosis and remodelling in heart failure: from basic mechanisms to early clinical development. Cardiovasc Res. 2022;118:3482-98.
61. Moazzam M, Zhang M, Hussain A, Yu X, Huang J, Huang Y. The landscape of nanoparticle-based siRNA delivery and therapeutic development. Mol Ther. 2024;32:284-312.
62. Yang L, Liu Z, Chen G, et al. MicroRNA-122-mediated liver detargeting enhances the tissue specificity of cardiac genome editing. Circulation. 2024;149:1778-81.
63. Shuvaev VV, Tam YK, Lee BW, et al. Systemic delivery of biotherapeutic RNA to the myocardium transiently modulates cardiac contractility in vivo. Proc Natl Acad Sci USA. 2025;122:e2409266122.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].